
Method Article
Interactome-Seq: A Protocol for Domainome Library Construction, Validation and Selection by Phage Display and Next Generation Sequencing
In This Article
Summary
The protocols described allow the construction, characterization and selection (against the target of choice) of a "domainome" library made from any DNA source. This is achieved by a research pipeline that combines different technologies: phage display, a folding reporter and next generation sequencing with a web tool for data analysis.
Abstract
Folding reporters are proteins with easily identifiable phenotypes, such as antibiotic resistance, whose folding and function is compromised when fused to poorly folding proteins or random open reading frames. We have developed a strategy where, by using TEM-1 β-lactamase (the enzyme conferring ampicillin resistance) on a genomic scale, we can select collections of correctly folded protein domains from the coding portion of the DNA of any intronless genome. The protein fragments obtained by this approach, the so called "domainome", will be well expressed and soluble, making them suitable for structural/functional studies.
By cloning and displaying the "domainome" directly in a phage display system, we have showed that it is possible to select specific protein domains with the desired binding properties (e.g., to other proteins or to antibodies), thus providing essential experimental information for gene annotation or antigen identification.
The identification of the most enriched clones in a selected polyclonal population can be achieved by using novel next-generation sequencing technologies (NGS). For these reasons, we introduce deep sequencing analysis of the library itself and the selection outputs to provide complete information on diversity, abundance and precise mapping of each of the selected fragment. The protocols presented here show the key steps for library construction, characterization, and validation.
Introduction
Here, we describe a high-throughput method for the construction and selection of libraries of folded and soluble protein domains from any genic/genomic starting source. The approach combines three different technologies: phage display, the use of a folding reporter and next generation sequencing (NGS) with a specific web tool for data analysis. The methods can be used in many different contexts of protein-based research, for identification and annotation of new proteins/protein domains, characterization of structural and functional properties of known proteins as well as definition of protein-interaction network.
Many open questions are still present in protein-based research and the development of methods for optimal protein production is an important need for several fields of investigation. For example, despite the availability of thousands of prokaryotic and eukaryotic genomes1, a corresponding map of the relative proteomes with a direct annotation of the coded proteins and peptides is still missing for the great majority of organisms. The catalogue of complete proteomes is emerging as a challenging goal requiring a huge effort in terms of time and resources. The gold standard for experimental annotation remains the cloning of all the Open Reading Frames (ORFs) of a genome, building the so called "ORFeome". Usually gene function is assigned based on homology to related genes of known activity but this approach is poorly accurate due to the presence of many incorrect annotations in the reference databases2,3,4,5. Moreover, even for proteins that have been identified and annotated, additional studies are required to achieve characterization in terms of abundance, expression patterns in different contexts, including structural and functional properties as well as interaction networks.
Furthermore, since proteins are composed of different domains, each of them showing specific features and differently contributing to protein functions, the study and the exact definition of these domains can allow a more comprehensive picture, both at the single gene and at the full genome level. All this necessary information makes protein-based research a wide and challenging field.
In this perspective, an important contribution could be given by unbiased and high-throughput methods for protein production. However, the success of such approaches, beside the considerable investment required, relies on the ability to produce soluble/stable protein constructs. This is a major limiting factor since it has been estimated that only about 30% of proteins can be successfully expressed and produced at sufficient levels to be experimentally useful6,7,8. An approach to overcome this limitation is based on the use of randomly fragmented DNA to produce different polypeptides, which together provide overlapping fragment representation of individual genes. Only a small percentage of the randomly generated DNA fragments are functional ORFs whilst the great majority of them are non-functional (due to the presence of stop codons inside their sequences) or encode for un-natural (ORF in a frame other than the original) polypeptides with no biological meaning.
To address all these issues, our group has developed an high-throughput protein expression and interaction analysis platform that can be used on a genomic scale9,10,11,12. This platform integrates the following techniques: 1) a method to select collections of correctly folded protein domains from the coding portion of DNA from any organism; 2) the phage display technology for selecting partners of interactions; 3) the NGS to completely characterize the whole interactome under study and identify the clones of interest; and 4) a web tool for data analysis for users without any bioinformatics or programming skills to perform Interactome-Seq analysis in an easy and user-friendly way.
The use of this platform offers important advantages over alternative strategies of investigation; above all the method is completely unbiased, high-throughput, and modular for study ranging from a single gene up to a whole genome. The first step of the pipeline is the creation of a library from randomly fragmented DNA under study, which is then deeply characterized by NGS. This library is generated using an engineered vector where genes/fragments of interest are cloned between a signal sequence for protein secretion into the periplasmic space (i.e., a Sec leader) and the TEM1 β-lactamase gene. The fusion protein will confer ampicillin resistance and the ability to survive under ampicillin pressure only if cloned fragments are in-frame with both these elements and the resulting fusion protein is correctly folded10,13,14. All clones rescued after antibiotic selection, the so called "filtered clones", are ORFs and, a great majority of them (more than 80%), are derived from real genes9. Moreover, the power of this strategy lies in the findings that all ORF filtered clones are encoding for correctly folded/soluble proteins/domains15. As many clones, present in the library and mapping in the same region/domain, have different starting and ending points, this allows unbiased, single-step identification of the minimum fragments that are likely to result in soluble products.
A further improvement in the technology is given by the use of NGS to characterize the library. The combination of this platform and of a specific web tool for data analysis gives important unbiased information on the exact nucleotide sequences and on the location of selected ORFs on the reference DNA under study without the need of further extensive analyses or experimental effort.
Domainome libraries can be transferred into a selection context and used as a universal instrument to perform functional studies. The high-throughput protein expression and interaction analysis platform that we integrated and that we called Interactome-Seq takes advantage of the phage display technology by transferring the filtered ORF into a phagemid vector and creating a phage-ORF library. Once re-cloned into a phage display context, protein domains are displayed on the surface of M13 particles; in this way domainome libraries can be directly selected for gene fragments encoding domains with specific enzymatic activities or binding properties, allowing interactome networks profiling. This approach was initially described by Zacchi et al.16 and later used in several other context13,17,18.
Compared to other technologies used to study protein-protein interaction (including yeast two hybrid system and mass spectrometry19,20), one major advantage is the amplification of the binding partner that occurs during phage display multiple rounds of selection. This increases the selection sensitivity thus allowing the identification of low abundant binding proteins' domains present in the library. The efficiency of the selection performed with ORF-filtered library is further increased due to the absence of non-functional clones. Finally, the technology allows the selection to be performed against both protein and non-protein baits21,22,23,24,25.
Phage selections using the domainome-phage library can be performed using antibodies coming from sera of patients with different pathological conditions, e.g. autoimmune diseases13, cancer or infection diseases as bait. This approach is used to obtain the so called "antibody signature" of the disease under study allowing to massively identify and characterize the antigens/epitopes specifically recognized by the patients' antibodies at the same time. Compared to other methods the use of phage display allows the identification of both linear and conformational antigenic epitopes. The identification of a specific signature could potentially have an important impact for understanding pathogenesis, new vaccine design, identification of new therapeutic targets and development of new and specific diagnostic and prognostic tools. Moreover, when the study is focused on infectious diseases, a major advantage is that the discovery of immunogenic proteins is independent from pathogen cultivation.
Our approach confirms that the folding reporters can be used on a genomic scale to select the "domainome": a collection of correctly folded, well expressed, soluble protein domains from the coding portion of the DNA and/or cDNA from any organism. Once isolated the protein fragments are useful for many purposes, providing essential experimental information for gene annotation as well as for structural studies, antibody epitope mapping, antigen identification, etc. The completeness of high-throughput data provided by NGS enables the analysis of highly complex samples, such as phage display libraries, and holds the potential to circumvent the traditional laborious picking and testing of individual phage rescued clones.
At the same time thanks to the features of the filtered library and to the extreme sensitivity and power of the NGS analysis, it is possible to identify the protein domain responsible of each interaction directly in an initial screen, without the need to create additional libraries for each bound protein. NGS allows to obtain a comprehensive definition of the whole domainome of any genic/genomic starting source and the data analysis web tool enables the obtainment of a highly specific characterization both from a qualitative and quantitative point of view of the interactome proteins' domains.
Protocol
1. Construction of the ORF Library (Figure 1)
- Preparation of insert DNA
- Fragments preparation from synthetic or genomic DNA
- Extract/purify DNA using standard methods26.
- Fragment DNA by sonication. If using a standard sonicator, as a general suggestion start with 30 s pulses at 100% power output.
NOTE: Pilot experiments should be done with different power and sonication times to set the optimal conditions for the DNA preparation. After each test determine the size of the DNA fragments by agarose gel electrophoresis. - Load the sonicated DNA onto 1.5% agarose gel, together with a 100 bp DNA ladder. Perform a short electrophoresis run at 5 V/cm for 15 min and cut the portion of the gel containing the smear of the fragmented DNA.
- Purify the insert DNA with a column-based gel extraction kit and measure the concentration using an UV spectrophotometer.
NOTE: At least 500 ng of purified inserts should be obtained after this step, to be ligated with 1 µg of digested vector, as described in step 1.3. Check quality of fragments preparation by evaluating A260nm/A280nm and A260nm/A230nm ratios since low quality of the sample will affect the ligation efficiency. - Treat up to 5 μg of the inserts with 1 μL of the Quick Blunting Kit enzyme mix, according to manufacturer's instructions. Inactivate enzymes by heating at 70 °C for 10 min. Samples can be stored at -20 °C until use.
- Fragments preparation from cDNA
- Extract RNA with standard methods (e.g., using TRizol or similar reagents).
- Fragment mRNA by heating prior to performing reverse transcription. The final DNA fragment length is controlled by mRNA boiling time and random primer concentration. For example, heat sample for 6 min at 95 °C.
- Prepare cDNA using random primers with any available kit following the manufacturer's protocol.
- Deplete cDNA of poly-dT tails by hybridization with biotinylated poly-dA for 3 h at 37 °C, and separate on streptavidin magnetic beads as described by Carninci et al.13
- Recover unbound material and purify with a column-based DNA purification kit following the manufacturer's instructions. Measure the concentration using an UV spectrophotometer. See Note in step 1.1.1.4.
- Fragments preparation from synthetic or genomic DNA
- Preparation of the filtering vector
- Digest 5 µg of purified cloning vector pFILTER312 with 10 U of EcoRV restriction enzyme, following manufacturer's protocol.
- Load 2 µL (200 ng) of the digested vector, together with 100 ng of the undigested vector and 1k bp molecular marker, on a 1% agarose gel, to check for proper digestion. Heat inactivate the restriction enzyme.
- Add 1/10 volume of 10x phosphatase buffer and 1 µL (5 U) of phosphatase and incubate at 37 °C for 15 min. Heat inactivate for 5 min at 65 °C.
- Purify digested plasmid by extraction from agarose gel, and measure the concentration using an UV spectrophotometer. Samples can be stored at -20 °C until use.
- Ligation and transformation
- Perform ligation as follows: for 1 µg of digested plasmid add 400 ng of phosphorylated inserts (plasmid:insert molar ratio 1:5), 10 µL of 10x Buffer for T4 DNA Ligase, 2 µL of high concentration T4 DNA ligase in a final volume of 100 µL. Incubate the reaction at 16 °C overnight. Heat inactivate at 65 °C for 10 min.
- Precipitate the ligation product by adding 1/10 volume of sodium acetate solution (3 M, pH 5.2) and 2.5 volumes of 100% ethanol. Mix and freeze at -80 °C for 20 min.
- Centrifuge at maximum speed for 20 min at 4 °C. Discard the supernatant.
- Add 500 µL of cold 70% ethanol to the pellet and centrifuge at maximum speed for 20 min at 4 °C. Discard the supernatant.
- Air dry the pellet. Resuspend the precipitated DNA into 10 µL of water.
- Perform bacterial cell electroporation.
NOTE: The use of high-efficiency cells (above 5x109 transformants per µg of DNA) is required. We suggest using Escherichia coli DH5αF' (F'/endA1 hsd17 (rK − mK+) supE44 thi-1recA1 gyrA (Nalr) relA1 (lacZYA-argF) U169 deoR (F80dlacD-(lacZ)M15) produced in house or purchased from several manufacturers.- Place appropriate number of microcentrifuge tubes and 0.1 cm-electroporation cuvettes on ice. Add 1 µL of purified ligation solution (in DI water) to 25 µL of the cells and flick the tube a few times.
- Transfer the DNA-cell mixture to the cold cuvette, tap on countertop 2x, wipe water from exterior of cuvette, place in the electroporation module and press pulse.
- Perform electroporation with a standard electroporator machine using 25 µF, 200 Ω and 1.8 kV. Time constant must be 4-5 ms.
- Immediately add 1 mL of liquid 2xYT medium without any antibiotic, transfer to a 10 mL tube and allow to grow at 37 °C, shaking at 220 rpm for 1 h.
- Plate transformed DH5αF' on 15 cm 2xYT agar plates supplemented with 34 µg/mL chloramphenicol (pFILTER resistance) and 25 µg/mL ampicillin (selective marker for ORFs) and incubate overnight at 30 °C.
- Plate dilutions of the library on 10 cm 2xYT agar plates supplemented with chloramphenicol + ampicillin and with chloramphenicol only, to perform library titration. Incubate overnight at 30 °C.
- pFILTER-ORF library validation
- Test 15-20 colonies from both chloramphenicol and chloramphenicol/ampicillin plates to estimate insert size distribution. Pick single colonies with a tip and dilute them separately in 100 µL of 2xYT medium without antibiotics. Use 0.5 µL of this solution as DNA template for a PCR reaction, with any standard TaqDNA polymerase following manufacturer's protocol.
- Perform 25 cycles of amplification using a T annealing of 55 °C and an extension time of 40 s at 72 °C. Primer sequences are provided in the Table of Materials.
- Load the PCR products on 1.5% agarose gel, together with a 100 bp DNA ladder and run.
- pFILTER-ORF library collection
- Collect bacteria from the 150 mm plates by adding 3 mL of fresh 2xYT medium and harvesting them with a sterile scraper, mix thoroughly, supplement them with 20% sterile glycerol and store at -80 °C in small aliquots.
- Purify plasmid DNA from one aliquot of the library (before the addition of glycerol) using a column-based plasmid extraction kit, following manufacturer's instructions.
- Measure the concentration with the UV spectrophotometer. Samples can be stored at -20 °C until be used for phagemid library preparation and/or characterization by NGS.
2. Subcloning of Filtered ORFs in a Phagemid Vector (Figure 2)
- Preparation of ORF filtered DNA fragments
- Set up restriction enzyme digestion of 5 µg of purified vector from the pFILTER-ORF library vector adding 10 U of BssHII and incubating as per manufacturer's protocol. Inactivate the enzyme and digest with 10 U of NheI.
- Load the digested DNA onto 1.5% agarose gel, together with a 100 bp DNA ladder. Perform a short electrophoresis run at 5 V/cm for 15 min or just enough to distinguish the smear of excised fragments and cut the portion of the gel containing them.
- Purify the insert DNA with a column-base gel extraction kit and measure the concentration using an UV spectrophotometer.
- Preparation of phagemid DNA
- Set up restriction enzyme digestion of 5 µg of purified pDAN527 as for the inserts.
- Purify digested plasmid by running digested DNA on a 0.75% agarose gel and extract from gel with a column-based kit.
- Measure the concentration using an UV spectrophotometer. Samples can be stored at -20 °C until use.
- Library ligation, transformation and collection
- Perform ligation and transformation as described for pFILTER vector.
- Plate transformed DH5αF' on 150 mm 2xYT agar plates supplemented with 100 µg/mL ampicillin and incubate overnight at 30 °C.
- Plate dilutions of the library on 100 mm 2xYT agar plates supplemented with 100 µg/mL ampicillin to determine the library size.
- Perform library validation by PCR of randomly picked clones as described in step 1.4.
- Collect the phagemid-ORF library by harvesting bacteria from 150 mm plates, mix thoroughly, supplement them with 20% sterile glycerol and store at -80 °C in small aliquots .
- Purify plasmid DNA from one aliquot of the library using a column-based plasmid extraction kit, following manufacturer's instructions.
- Measure the concentration at the UV spectrophotometer. Samples can be stored at -20 °C until be used for characterization by NGS.
3. Phage Library Preparation and Selection Procedure
- Phage production
- Dilute a stock aliquot of the phagemid library into 10 mL of 2xYT liquid broth supplemented with 100 µg/mL ampicillin in order to have an OD600nm = 0.05.
- Grow the diluted library in a sterile flask 5-10 times bigger than the original volume, at 37 °C with shaking at 220 rpm until reaches OD600nm = 0.5.
- Infect bacteria with helper phage (e.g. M13K07) at a multiplicity of infection 20:1. Leave at 37 °C for 45 min with occasional agitation (every 10 min).
- Centrifuge bacteria at 4000 x g for 10 min at room temperature. Discard supernatant, re-suspend bacteria pellet in 40 mL of 2xYT liquid broth supplemented with 100 µg/mL ampicillin and 50 µg/mL kanamycin and grow at 28 °C with shaking at 220 rpm for overnight.
- The day after, centrifuge bacteria at 4000 x g for 20 min at 4 °C. Collect the supernatant containing phages.
- PEG-precipitation of phages.
- Add 1/5 volume of a 0.22 µm filtered PEG/NaCl solution (20% w/v PEG 6000, 2.5 M NaCl) to the cleared phages and incubate on ice for 30-60 min.
NOTE: Solution became smoky after few minutes, indicating a successful phage precipitation. The cloudiness of the solution will increase over the incubation time. - Centrifuge at 4000 x g for 15 min at 4 °C. A white small pellet of phages will form.
- Resuspend it in 1 mL of sterile PBS. Transfer to 1.5 mL tube and centrifuge at 4 °C for 10 min at maximum speed to remove contaminant bacteria. A brown pellet will form.
- Transfer supernatant containing phages to a new tube. Keep phages on ice for successive titration and phage selection.
- Add 1/5 volume of a 0.22 µm filtered PEG/NaCl solution (20% w/v PEG 6000, 2.5 M NaCl) to the cleared phages and incubate on ice for 30-60 min.
- Phage titration
- Prepare serial dilutions of the phage solution. Put 10 µL of the phage solution in 990 µL of PBS to obtain 10-2 dilution. Dilute again this preparation to make 10-4 and from this obtain a 10-6 dilution.
- Grow DH5αF' bacteria cells into 2xYT liquid medium at 37 °C with shaking until OD600nm = 0.5 is reached. Transfer 1 mL of the prepared bacteria into 1.5 mL tube and immediately infect with 1 µL of the 10-4 phage dilution. Incubate without shaking at 37 °C for 45 min. Repeat the same procedure for the 10-6 dilution.
- Plate dilutions of the infected bacteria into 100 mm 2xYT plate. Put the plate at 30 °C overnight.
- Plate 100 µL of uninfected DH5αF' on a 2xYT agar plate supplemented with 100 µg/mL ampicillin to check the absence of contamination in the preparation.
- The day after count the number of colonies and calculate the phage titer. Express titer as number of phages/mL. Expected titer is 1012-13 phages/mL.
- Phage selection
- Phage selection using as bait purified antibodies
- Saturate phages by diluting 200 µL of phage preparation into an equal volume of PBS-4% skimmed milk and incubate for 1 h at room temperature in slow rotation. This step allows blocking of phages for unspecific binding. Transfer 30 µL of protein-G coated magnetic beads to a 1.5 mL tube.
- Wash two times as follows: add 500 µL of PBS, incubate on a wheel in slow rotation for 2 min at room temperature, draw the beads to one side of the tube using a magnet and remove the supernatant.
- Incubate saturated phages with washed beads for 30 min at room temperature with slow rotation.
- Draw the beads to one side using a magnetic field. Collect the supernatant containing phages to be used for the selection step.
- Prepare magnetic beads while performing the previous step, by conjugating the purified antibodies. Wash 30 µL of protein-G coated magnetic beads as described above. Dilute 10 µg of purified antibodies in 500 µL of PBS, add to the washed beads and incubate in slow rotation at room temperature for 45 min. Wash twice with PBS.
NOTE: Perform two different preparations of magnetic beads: one with antibodies of interest and one with control antibodies, e.g., antibodies purified from healthy donors. The sequence of antigens selected with control antibodies are subtracted during the analysis step of the outputs. Alternatively, magnetic beads loaded with control antibodies can be used to perform a pre-clearing step of the phages (follow the protocol for the incubation with un-conjugated beads). - Phage selection: draw beads to one side of the tube using a magnet, remove the last wash, add phages and incubate with slow rotation at room temperature for 90 min. Wash 5 times with 500 µL of PBS-0.1% Tween-20 and 5 times with PBS.
- Elute bound phages, representing the output of the selection, by mixing the beads with 1 mL of DH5αF' cells grown at OD600 = 0.5. Incubate bacteria with beads for 45 min at 37 °C with occasional shaking (every 10 min). Plate the output on a 150 mm 2xYT agar plate supplemented with 100 µg/mL ampicillin.
- Plate 100 µL of undiluted and of different dilutions of the output (10-1 to 10-5) to perform titration. The day after collect bacteria from the 150 mm plates by adding 3 mL of fresh 2xYT medium and harvesting them with a sterile scraper, mix thoroughly, supplement them with 20% sterile glycerol and store at -80 °C in small aliquots.
- Grow one aliquot again to perform a second round of selection. Repeat all the panning procedure as described above except for the washing conditions. In this case wash 10 times with PBS-1% Tween-20 (pour solution in the tube and pour out again immediately). Then add 500 µL of PBS and incubate on rotation at room temperature for 10 min. Perform other 10 washes with PBS. Proceed with the elution step as for the first round of selection.
- Extract plasmid DNA from one aliquot of the output using a column-based kit, following manufacturers' instructions. Store plasmid at -20 °C until it will be used for deep sequencing.
- Phage selection using as bait recombinant proteins
- Saturate phages by diluting 200 µL of phage preparation into an equal volume of PBS-4% skimmed milk and incubate for 1 h at room temperature in slow rotation.
- Add 100 µL of streptavidin magnetic beads. Incubate for 1 h at room temperature to select streptavidin-binding phages. Remove the streptavidin-bound phages by drawing the beads to one side using a magnet. Take supernatant from the previous step and add biotinylated protein (in a concentration of 100-550 nM) and incubate on a rotor at room temperature for 30 min to 1 h.
- Prepare magnetic beads: while performing the previous step, wash 100 µL of streptavidin-magnetic beads with PBS, resuspend in PBS 2% skimmed milk and incubate with rotation at room temperature for 30 min to 1 h.
- Phage selection: Draw beads to one side of the tube using a magnet, remove PBS-2% milk and resuspend beads with phage-protein mix. Incubate with slow rotation at room temperature for 90 min.
- Draw beads to one side of the tube using a magnet, discard the supernatant and wash them carefully five times with 500 µL of PBS 0.1% Tween-20. Perform elution as described in the previous session.
- Phage selection using as bait purified antibodies
4. Phage Library Deep Sequencing Platform (Figure 3)
- DNA inserts recovery from pFILTER-ORF-library, pDAN5-ORF-library or selected-phage-libraries
- Thaw one aliquot of the library, quantify it by using a spectrophotometer, recover DNA inserts by amplification with specific primers.
NOTE: The primers used to rescue the inserts are linked at their 5' end to adaptors sequences, thus allowing the successive indexing of the amplicon pools obtained and the direct sequencing of the DNA inserts recovered by using the sequencers. Their sequence is in the Table of Materials. The adaptors are indicated in bold, and the specific primers are indicated in italics. - Use 2.5 µL of the (pFILTER/phagemid/selected-phage) library as DNA template for a PCR reaction.
- Use the following program: 95 °C for 3 min; 25 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s; 72 °C for 5 min. Hold at 4 °C.
NOTE: At this point it is recommended to run 1 µL of the PCR product on a Bioanalyzer or TapeStation to verify the size of the amplicons and check that they are in the correct range.
- Thaw one aliquot of the library, quantify it by using a spectrophotometer, recover DNA inserts by amplification with specific primers.
- PCR clean-up
- Bring the magnetic beads (e.g., AMPure) to room temperature. Transfer the entire PCR product from the PCR tube to a 1.5 mL tube. Vortex the magnetic beads for 30 s to make sure that the beads are evenly dispersed. Add 20 µL of magnetic beads to each tube containing the PCR product, mix by gently pipetting. Incubate at room temperature without shaking for 5 min.
- Place the plate on a magnetic stand for 2 min or until the supernatant has cleared. With the PCR products on the magnetic stand, remove and discard the supernatant.
- Wash the beads with freshly prepared 80% ethanol, with the PCR products on the magnetic stand, as follows: add 200 µL of freshly prepared 80% ethanol to each sample well; incubate the plate on the magnetic stand for 3 s; carefully remove and discard the supernatant.
- Perform a second ethanol wash, with the PCR products on the magnetic stand; at the end of second wash carefully remove all the ethanol and allow the beads to air-dry for 10 min.
- Remove the PCR products from the magnetic stand, add 17.5 µL of 10 mM Tris pH 8.5 to each tube, gently pipette up and down 10 times, make sure that beads are fully resuspended. Incubate at room temperature for 2 min.
- Place the tube on the magnetic stand for 2 min or until the supernatant has cleared, carefully transfer 15 µL of the supernatant containing the purified PCR products to a new 1.5 mL tube. Store the purified PCR products at -15 ° C to -25 °C for up to a week if you do not immediately proceed to Index PCR.
- Index PCR
NOTE: After PCR clean up, perform Index PCR. Use the Nextera XT Index kit; thus it will be possible to sequence the resulting double indexed libraries within multiplexed Illumina runs.- Transfer all the 15 μL containing each product purified into a new PCR tube and set up the following reaction containing: 15 µL of purified amplicon product, 5 µL of Index Primer 1 and 5 µL of Index Primer 2, 25 µL of 2x PCR mix; final volume of 50 µL.
- Perform PCR on a thermal cycler using the following program: 95 °C for 3 min, 8 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s; 72°C for 5 min, then hold at 4 °C.
- PCR clean-up 2
- Follow the same protocol described in the section 4.2 for PCR clean up with the following changes: in the first step add 56 µL of magnetic beads to each 50 µL of PCR product.
- Resuspend the beads in 27.5 µL of 10 mM Tris pH 8.5 in the final step of purification and transfer 25 µL to a new tube (this is the purified final library ready for quantification and then sequencing).
- Store the plate at -15 °C to -25 °C for up to a week if not proceeding to Library Quantification.
- Qualitative and quantitative evaluation of the sequencing library
- After purification, run 1 µL of a 1:10 dilution of the final library on a bioanalyzer to verify the size and quantify it selecting the region of the final library trace.
- In parallel perform the library quantification by Real Time PCR by using a library quantification kit per manufacturer's protocol.
- Libraries sequencing
- Pool the dual indexed libraries produced together with other dual indexed sequencing libraries. Sequence this kind of library by generating long reads, at least 250 bp paired end by using both the Hiseq2500 or the MiSeq instruments to obtain in the first case 250bp PE reads and in the second case 300 bp PE reads.
5. Bioinformatic Data Analysis by Using the Interactome-Seq Web Tool
- Analyze the reads originated from pFILTER/phagemid/selected-phage library sequencing with the Interactome-seq data analysis pipeline. The web tool is freely available at the following address: http://interactomeseq.ba.itb.cnr.it/
Results
The filtering approach is schematized in Figure 1. Each kind of intronless DNA can be used. In Figure 1A the first part of the filtering approach is represented: after loading on an agarose gel or a bioanalyzer, a good fragmentation of the DNA of interest appears as a smear of fragments with a length distribution in the desired size of 150-750 bp. A representative virtual gel image of the fragmented DNA obtained is given. Fragments loaded on the agarose gel are then recovered, end-repaired and phosphorylated, and then cloned into a previously blunted pFILTER vector to create a library of random DNA fragments. Performing each step of the cloning procedure under optimal conditions is required to obtain good quality library with a total coverage of the DNA under study.
In Figure 1B the filtering approach is represented: the library is grown in the presence of chloramphenicol (pFILTER resistence) alone or chloramphenicol and ampicillin to select for ORF-containing colonies. Only colonies having a DNA fragment corresponding to an ORF produce a functional β-lactamase and survive when antibiotic selection is present. Figure 1C shows how increasing selective pressure allows selection of good folder ORFs versus poor folder ones. The expected result is a decrease of the library size of about 20-fold. Higher number of surviving clones indicates insufficient selective pressure.
ORF fragments can be easily recovered from the filtered library for subsequent application; for interaction studies our strategy takes advantage of phage display technology. In Figure 2, the principal steps of phage library construction are represented: an adequate library is prepared by cutting out filtered fragments from the pFILTER vector and re-cloning into a phagemid plasmid in fusion with the sequence coding for the phage capside protein g3p. Once infected with helper phage, the presence of the vector into bacteria cells allows the production of phage particles displaying ORF-g3p fusion products on their surface thus making the filtered library available for phage display selection and further analysis.
All the libraries are deeply analyzed by NGS, as well as the outputs of the phage selections, as shown in the second part of Figure 3. DNA fragments are rescued from growing colonies by PCR amplification with specific oligonucleotides annealing on the plasmid backbone and carrying specific adapters for the sequencing. NGS is performed and reads are then analyzed with the Interactome-Seq data analysis web tool.
In Figure 4 we reported a schematic representation of the selection procedure of an ORF filtered phage display library. The selection in this example is performed by using antibodies present in the sera from patients affected by different pathologies (i.e. infective pathologies, autoimmune pathologies, cancer). In this case the phage library directly interacts with the antibodies present in the patients' sera and in this way putative specific antigens can be enriched because they are recognized by disease specific antibodies. In this kind of experiment, usually the library is also selected using control sera from healthy patients in order to have a background signal to be used for successive comparison and normalization procedures.
Selections are performed using sera from the same type of patients usually grouped together into different pools in order to reduce inter-individual variability of sera antibody titer. Each pool is independently used for two to three consecutive rounds of selection, to enrich the library for immune-reactive clones specific for the pathology under study. Test set antibodies are incubated with library phages, immune-complexes are recovered by protein A coated magnetics-beads and bound phages are eluted by standard procedures. The selection cycles are performed with increasing washing and binding stringency.
The reads generated by NGS can be analyzed using the Interactome-Seq web tool specifically developed to manage this kind of data. Interactome-Seq data analysis workflow is composed of four sequential steps that, starting from raw sequencing reads, generates the list of putative domains with genomic annotations (Figure 5A). In the first step INPUT (Figure 5A - red box), Interactome-Seq checks if the input files (raw reads, reference genome sequence, annotation list) are properly formatted. In the second step PREPROCESSING (Figure 5A - orange box), low-quality sequencing data are first trimmed using Cutadapt28 depending on quality scores and reads with less than 100 bases in length are discarded. In a subsequent READ ALIGNMENT step (Figure 5A - green box), the remaining reads are aligned with blastn29 to the genome sequence allowing up to 5% of mismatches. A SAM file is generated and only reads with quality score greater than 30 (Q>30) are processed using SAMtools30 and converted into a BAM file. After alignment, Interactome-Seq performs the DOMAINS DETECTION (Figure 5A - blue box), invoking Bedtools31 to filter reads overlapping at least for 80% of their length inside transcripts; the coverage, max depth and focus values are then calculated for each ORF portion covered by mapping reads. The coverage represents the total number of reads assigned to a gene; the depth is the maximum number of reads covering a specific genic portion; the focus is an index obtained from the ratio between max depth and coverage, and it can range between 0 and 1. When the focus is higher than 0.8 and the coverage is higher than the average coverage observed for all mapping regions in the BAM file, the CDS portion is classified as a putative domain/epitope. The last step of the Interactome-Seq pipeline is the OUTPUT (Figure 5A - violet box), a list of putative domains is generated in tabular separated format. The Interactome-Seq pipeline has been included in a web-tool to enable users without any bioinformatics or programming skills to perform Interactome-Seq analysis through the graphical interface and to obtain their results in an easy and user-friendly format. As shown in Figure 5B, the output results of an analysis are displayed using JBrowse32 to enable visualization and exploration. Interactome-Seq generates tracks in the genome browser corresponding to putative domains detected and provides also classical Venn diagrams to show intersections between common putative domains enriched for example in different selections experiments.

Figure 1: Schematic overview of the main steps for the construction of the ORF-filtering library
A) DNA from different source is sonicated and fragmented into random fragments of 150-750 bp length. Fragments are recovered from gel and cloned as blunt into the pFILTER vector; B) filtering step using β-lactamase as a folding reporter. Vector containing not ORF fragments are negatively selected on ampicillin while ORF cloned fragments allow colonies to grow; C) application of an increasing selective pressure (ampicillin concentration in solid growth media from 0 to >100 μg/mL) allow selection of better folded fragments. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Schematic overview of the main steps for the construction of the phage library
A) ORF-filtered fragments are cut out from the filtered vector using specific restriction enzymes. After recovery and purification, fragments are cloned into phagemid vector and transformed; B) phagemid bacterial library is infected with helper phage and, after overnight growth, phages are PEG-precipitated and collected. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: ORF libraries sequencing
Sequencing is performed on both the original ORF selected library as well as on the phage display library; 1) on both cases colonies grown are recovered and DNA extracted; 2) DNA fragments are recovered by amplification using specific primers linked to adaptors for sequencing; 3-4) fragments are recovered and deep sequenced using NGS; 5) data are analyzed by using the Interactome-Seq pipeline. Please click here to view a larger version of this figure.
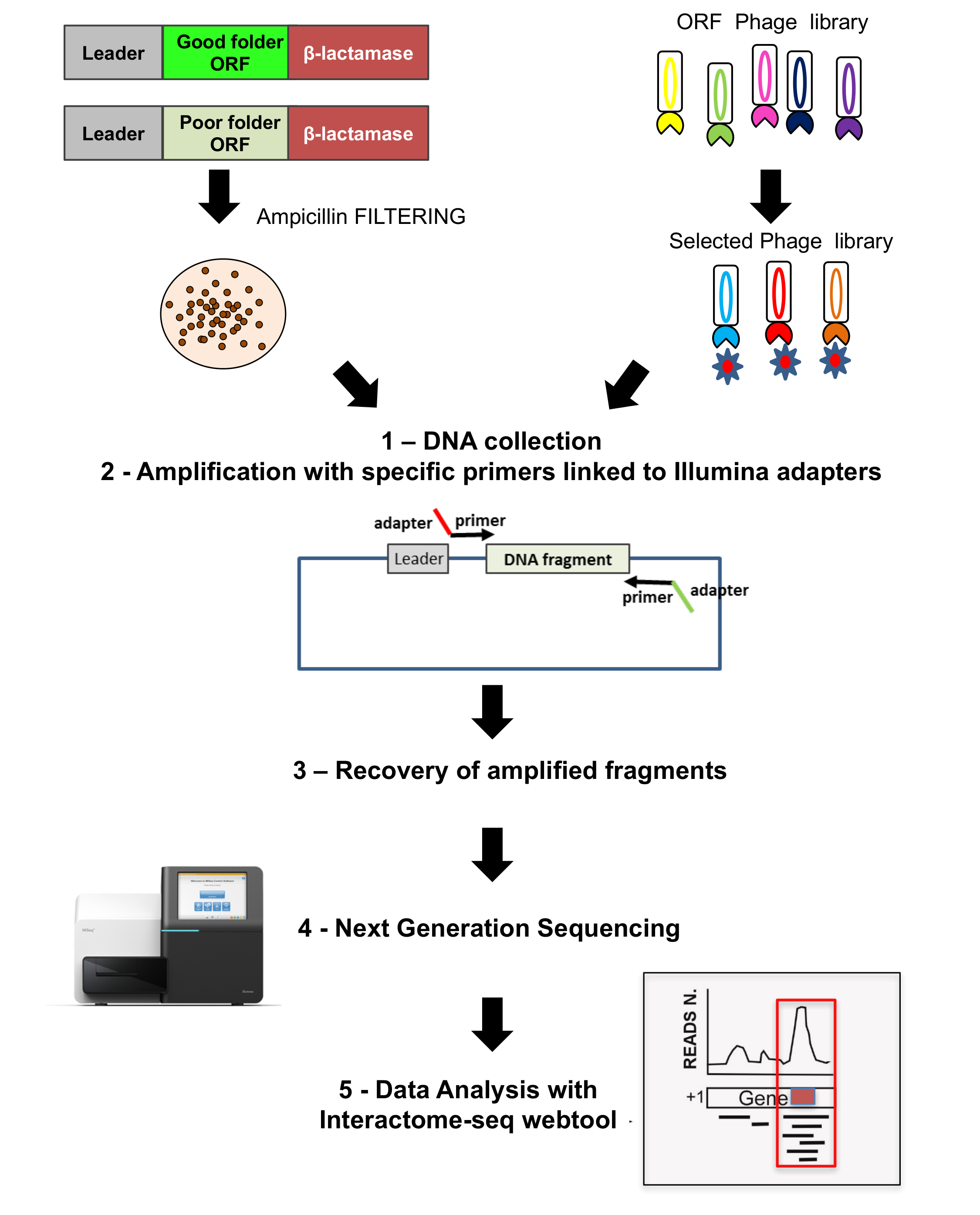{kind=link}

Figure 4: Schematic overview of library selection using patients' antibodies
Phage library is used for selection against antibodies from patients' sera. Antibodies are immobilized on magnetic beads, the phage library capture/selection is performed, three cycles of washes are performed and afterwards selected phages are recovered and used to re-infect E. coli. Re-infected E. coli cells are plated in selective pressure (ampicillin 100 μg/mL). ORF fragments are recovered by amplification and amplicon pools are then sequenced by NGS. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Schematic overview of library analysis
A) Representation of the data analysis workflow, starting from raw FASTQ files to the final annotated domains lists; B) schematic representation of the inputs and outputs of the Interactome-Seq web tool. Please click here to view a larger version of this figure.
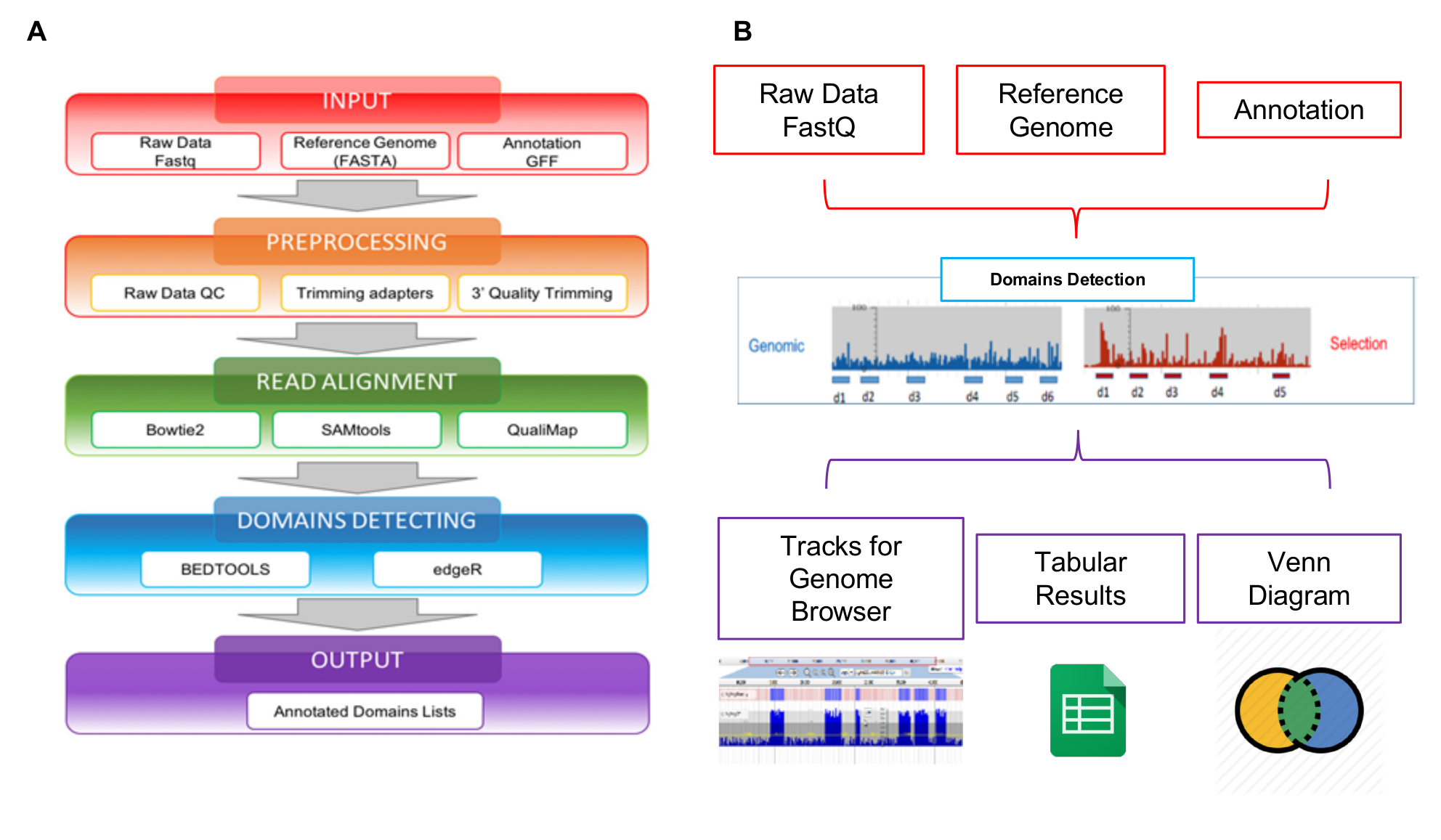{kind=link}
Discussion
The creation of a high quality highly diverse ORFs filtered library is the first critical step in the whole procedure since it will affect all the subsequent steps of the pipeline.
An important advantageous feature of our method is that any source of (intronless) DNA (cDNA, genomic DNA, PCR derived or synthetic DNA) is suitable for library construction. The first parameter that should be taken into account is that the length of the DNA fragments cloned into the pFILTER vector should provide a representation of the entire collection of the domains of a genome or a transcriptome, the so called "domainome". We have demonstrated that protein domains can be successfully cloned, selected and finally identified starting from DNA fragments with a length distribution spanning from 150 to 750 bp33,34, and this is in line with what is reported in the literature showing that most protein domains are of 100 aa length (with a range from 50 to 200 aa)15.
DNA starting material must be fragmented into the size range of choice and later cloned into the filtering (pFILTER)12 vector. During these steps, potential bias could be avoided maximizing the efficiency of all the cloning steps reactions included in the protocol, in particular fragment end-repairing and phosphorylation. The vector preparation is challenging and should be made under optimal conditions as well, to avoid both plasmid degradation and/or contamination by undigested vector.
Once the library has been created, it should be "filtered" in order to retain only ORFs folded fragments. A key parameter to modulate this step is the selective pressure applied that can be modified according to the stringency of the filtering desired. Selection is performed using ampicillin: the higher the concentration used, the lower the number of transformed bacteria colonies able to survive. This reflects the ability of the filtering method to select for good- versus poor- folder ORFs34. This reduction in the number of clones is balanced by the increase in folding properties of selected fragments. Usually, the ampicillin concentration should be enough to reduce to about 1/20 the number of bacterial colonies with respect to those that could be obtained growing the library on chloramphenicol only.
Library validation is usually done by PCR amplification of randomly picked colonies and their sequencing. PCR amplification of some colonies is suggested in order to have a quick estimation of the quality of the library: the length of the inserts should be in the expected range of 150-750 bp and different colonies should present inserts with different size indicating good library preparation in term of variability. This conventional strategy of screening, when applied as the only method for library validation, is not comprehensive and is time consuming, allowing the analysis of only a limited number of colonies and having a high chance of missing most of the important clones. Our approach is based on deep sequencing of the library, this provides complete information on library diversity and abundance and precise mapping of each of the selected fragments.
The implementation of NGS technology with the filtering approach increases the deepness of the analysis by several orders of magnitude. Recently, we have optimized the protocol for sequencing the ORF libraries by using the Illumina platform, and developed a specific web tool for data analysis that makes the analysis of these kind of data for every user without any bioinformatics programming skills.
The library "per se" is a "universal instrument" and can be exploited in different contexts for protein expression and/or selection. Our methodological approach is based on the transferring of the produced ORFeome into a phage display context. Protein fragments are expressed on the phage surface and became suitable for subsequent selection.
This is made by rescuing the filtered ORFs from the pFILTER library by digestion with specific restriction enzymes and re-cloning them into a compatible phagemid vector allowing their fusion with the phage protein g3p.
After the phagemid-ORF library is created, it can be used for the selection against different targets, such as a putative binding protein10 or purified antibodies35,36 as described here. Since phage particles will display on their surface the filtered ORFs, this results in a much more effective selection procedure due to the absence of non-displaying clones that usually overtake it.
After the selection of the phage display ORF library, the output clones can be sequenced and analyzed with the same pipeline. NGS can provide a complete and statistically significant ranking of the most frequently selected ORFs and this allows the identification of the proteins mostly interacting with the bait used. Given the presence of many different versions of each domain differing by few amino acids, the overlap between different sequenced clones also identifies the minimum fragment/domain showing binding properties. Finally, thanks to the coupling of genotype and phenotype information into the phage library, once the domains of choice have been identified, the DNA sequence can be easily rescued from the library for further studies, in vitro and in vivo validation and characterization.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by a grant from the Italian Ministry of Education and University (2010P3S8BR_002 to CP).
Materials
| Name | Company | Catalog Number | Comments |
| Sonopuls ultrasonic homogenizer | Bandelin | HD2070 | or equivalent |
| GeneRuler 100 bp Plus DNA Ladder | Thermo Scientific | SM0321 | or equivalent |
| GeneRuler 1 kb DNA Ladder | Thermo Fisher Scientific | SM0311 | or equivalent |
| Molecular Biology Agarose | BioRad | 161-3102 | or equivalent |
| Green Gel Plus | Fisher Molecular Biology | FS-GEL01 | or equivalent |
| 6x DNA Loading Dye | Thermo Fisher Scientific | R0611 | or equivalent |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | or equivalent |
| Quick Blunting Kit | New England Biolabs | E1201S | |
| NanoDrop 2000 UV-Vis Spectrophotometer | Thermo Fisher Scientific | ND-2000 | |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | 4368813 | |
| Streptavidin Magnetic Beads | New England Biolabs | S1420S | or equivalent |
| QIAquick PCR purification Kit | Qiagen | 28104 | or equivalent |
| EcoRV | New England Biolabs | R0195L | |
| Antarctic Phosphatase | New England Biolabs | M0289S | |
| T4 DNA Ligase | New England Biolabs | M0202T | |
| Sodium Acetate 3M pH5.2 | general lab supplier | ||
| Ethanol for molecular biology | Sigma-Aldrich | E7023 | or equivalent |
| DH5aF' bacteria cells | Thermo Fisher Scientific | ||
| 0,2 ml tubes | general lab supplier | ||
| 1,5 ml tubes | general lab supplier | ||
| 0,1 cm electroporation cuvettes | Biosigma | 4905020 | |
| Electroporator 2510 | Eppendorf | ||
| 2x YT medium | Sigma-Aldrich | Y1003 | |
| Ampicillin sodium salt | Sigma-Aldrich | A9518 | |
| Chloramphenicol | Sigma-Aldrich | C0378 | |
| DreamTaq DNA Polymerase | Thermo Fisher Scientific | EP0702 | |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | N0447S | |
| 96-well thermal cycler (with heated lid) | general lab supplier | ||
| 150 mm plates | general lab supplier | ||
| 100 mm plates | general lab supplier | ||
| Glycerol | Sigma-Aldrich | G5516 | |
| BssHII | New England Biolabs | R0199L | |
| NheI | New England Biolabs | R0131L | |
| QIAprep Spin Miniprep Kit | Qiagen | 27104 | or equivalent |
| M13KO7 Helper Phage | GE Healthcare Life Sciences | 27-1524-01 | |
| Kanamycin sulfate from Streptomyces kanamyceticus | Sigma-Aldrich | K1377 | |
| Polyethylene glycol (PEG) | Sigma-Aldrich | P5413 | |
| Sodium Cloride (NaCl) | Sigma-Aldrich | S3014 | |
| PBS | general lab supplier | ||
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher Scientific | 10003D | or equivalent |
| MagnaRack Magnetic Separation Rack | Thermo Fisher Scientific | CS15000 | or equivalent |
| Tween 20 | Sigma-Aldrich | P1379 | |
| Nonfat dried milk powder | EuroClone | EMR180500 | |
| KAPA HiFi HotStart ReadyMix | Kapa Biosystems, Fisher Scientific | 7958935001 | |
| AMPure XP beads | Agencourt, Beckman Coulter | A63881 | |
| Nextera XT dual Index Primers | Illumina | FC-131-2001 or FC-131-2002 or FC-131-2003 or FC-131-2004 | |
| MiSeq or Hiseq2500 | Illumina | ||
| Spectrophotomer | Nanodrop | ||
| Agilent Bioanalyzer or TapeStation | Agilent | ||
| Forward PCR primer | general lab supplier | 5’ TACCTATTGCCTACGGCA GCCGCTGGATTGTTATTACTC 3’ | |
| Reverse PCR primer | general lab supplier | 5’ TGGTGATGGTGAGTACTA TCCAGGCCCAGCAGTGGGTTTG 3’ | |
| Forward primer for NGS | general lab supplier | 5’ TCGTCGGCAGCGTCAGA TGTGTATAAGAGACAGGCA GCAAGCGGCGCGCATGC 3’; | |
| Reverse primer for NGS | general lab supplier | 5’ GTCTCGTGGGCTCGGAGA TGTGTATAAGAGACAGGGG ATTGGTTTGCCGCTAGC 3’; |
References
- Loman, N. J., Pallen, M. J. Twenty years of bacterial genome sequencing. Nat Rev Microbiol. 13 (12), 787-794 (2015).
- Jones, C. E., Brown, A. L., Baumann, U. Estimating the annotation error rate of curated GO database sequence annotations. BMC Bioinformatics. 8 (1), 170 (2007).
- Andorf, C., Dobbs, D., Honavar, V. Exploring inconsistencies in genome-wide protein function annotations: a machine learning approach. BMC Bioinformatics. 8 (1), 284 (2007).
- Wong, W. -. C., Maurer-Stroh, S., Eisenhaber, F. More Than 1,001 Problems with Protein Domain Databases: Transmembrane Regions, Signal Peptides and the Issue of Sequence Homology. PLoS Comput Biol. 6 (7), e1000867 (2010).
- Bioinformatics, B., et al. Identification and correction of abnormal, incomplete and mispredicted proteins in public databases. BMC Bioinformatics. 9 (9), (2008).
- Phizicky, E., Bastiaens, P. I. H., Zhu, H., Snyder, M., Fields, S. Protein analysis on a proteomic scale. Nature. 422 (6928), 208-215 (2003).
- DiDonato, M., Deacon, A. M., Klock, H. E., McMullan, D., Lesley, S. A. A scaleable and integrated crystallization pipeline applied to mining the Thermotoga maritima proteome. J Struct Funct Genomics. 5 (1-2), 133-146 (2004).
- Nordlund, P., et al. Protein production and purification. Nat Methods. 5 (2), 135-146 (2008).
- Zacchi, P., Sblattero, D., Florian, F., Marzari, R., Bradbury, A. R. M. Selecting open reading frames from DNA. Genome Res. 13 (5), 980-990 (2003).
- Di Niro, R., et al. Rapid interactome profiling by massive sequencing. Nucleic Acids Res. 38 (9), e110 (2010).
- Gourlay, L. J., et al. Selecting soluble/foldable protein domains through single-gene or genomic ORF filtering: Structure of the head domain of Burkholderia pseudomallei antigen BPSL2063. Acta Crystallogr Sect D Biol Crystallogr. 71 (Pt 11), 2227-2235 (2015).
- D'Angelo, S., et al. Filtering "genic" open reading frames from genomic DNA samples for advanced annotation. BMC Genomics. 12 (Suppl 1), S5 (2011).
- D'Angelo, S., et al. Profiling celiac disease antibody repertoire. Clin Immunol. 148 (1), 99-109 (2013).
- Robinson, M. D., McCarthy, D. J., Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26 (1), 139-140 (2009).
- Heger, A., Holm, L. Exhaustive enumeration of protein domain families. J Mol Biol. 328 (3), 749-767 (2003).
- Zacchi, P., Sblattero, D., Florian, F., Marzari, R., Bradbury, A. R. M. Selecting open reading frames from DNA. Genome Res. 13 (5), 980-990 (2003).
- Faix, P. H., Burg, M. A., Gonzales, M., Ravey, E. P., Baird, A., Larocca, D. Phage display of cDNA libraries: Enrichment of cDNA expression using open reading frame selection. Biotechniques. 36 (6), 1018-1029 (2004).
- Patrucco, L., et al. Identification of novel proteins binding the AU-rich element of α-prothymosin mRNA through the selection of open reading frames (RIDome). RNA Biol. 12 (12), 1289-1300 (2015).
- Collins, M. O., Choudhary, J. S. Mapping multiprotein complexes by affinity purification and mass spectrometry. Curr Opin Biotechnol. 19 (4), 324-330 (2008).
- Suter, B., Kittanakom, S., Stagljar, I. Two-hybrid technologies in proteomics research. Curr Opin Biotechnol. 19 (4), 316-323 (2008).
- Nakai, Y., Nomura, Y., Sato, T., Shiratsuchi, A., Nakanishi, Y. Isolation of a Drosophila gene coding for a protein containing a novel phosphatidylserine-binding motif. J Biochem. 137 (5), 593-599 (2005).
- Deng, S. J., et al. Selection of antibody single-chain variable fragments with improved carbohydrate binding by phage display. J Biol Chem. 269 (13), 9533-9538 (1994).
- Danner, S., Belasco, J. G. T7 phage display: A novel genetic selection system for cloning RNA-binding proteins from cDNA libraries. Proc Natl Acad Sci. 98 (23), 12954-12959 (2001).
- Gargir, A., Ofek, I., Meron-Sudai, S., Tanamy, M. G., Kabouridis, P. S., Nissim, A. Single chain antibodies specific for fatty acids derived from a semi-synthetic phage display library. Biochim Biophys Acta - Gen Subj. 1569 (1-3), 167-173 (2002).
- Patrucco, L., et al. Identification of novel proteins binding the AU-rich element of α-prothymosin mRNA through the selection of open reading frames (RIDome). RNA Biol. 12 (12), 1289-1300 (2015).
- Ausubel, F. M., et al. Current Protocols in Molecular Biology. Mol Biol. 1 (2), 146 (2003).
- Sblattero, D., Bradbury, A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat Biotechnol. 18, 75-80 (2000).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 17 (1), 10 (2011).
- Camacho, C., et al. BLAST+: architecture and applications. BMC Bioinformatics. 10 (1), 421 (2009).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Quinlan, A. R. BEDTools: The Swiss-Army tool for genome feature analysis. Curr Protoc Bioinforma. , (2014).
- Skinner, M. E., Uzilov, A. V., Stein, L. D., Mungall, C. J., Holmes, I. H. JBrowse: A next-generation genome browser. Genome Res. 19 (9), 1630-1638 (2009).
- Gourlay, L. J., et al. Selecting soluble/foldable protein domains through single-gene or genomic ORF filtering: Structure of the head domain of Burkholderia pseudomallei antigen BPSL2063. Acta Crystallogr Sect D Biol Crystallogr. 71, 2227-2235 (2015).
- D'Angelo, S., et al. Filtering "genic" open reading frames from genomic DNA samples for advanced annotation. BMC Genomics. 12 (Suppl 1), S5 (2011).
- Di Niro, R., et al. Characterizing monoclonal antibody epitopes by filtered gene fragment phage display. Biochem J. 388 (Pt 3), 889-894 (2005).
- D'Angelo, S., et al. Profiling celiac disease antibody repertoire. Clin Immunol. 148 (1), 99-109 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved