
Method Article
Interactoma-Seq: Un protocollo per la costruzione della libreria Domainome, la convalida e la selezione di Phage Display e sequenziamento di nuova generazione
In questo articolo
Riepilogo
I protocolli descritti consentono la costruzione, la caratterizzazione e la selezione (contro il bersaglio di scelta) di una libreria di "domainome" fatta da qualsiasi fonte di DNA. Questo risultato è ottenuto da una pipeline di ricerca che combina diverse tecnologie: phage display, un reporter pieghevole e sequenziamento di nuova generazione con uno strumento web per l'analisi dei dati.
Abstract
Pieghevole reporter sono proteine con fenotipi facilmente identificabili, quali resistenza agli antibiotici, cui pieghevole e la funzione è compromessa quando fusa a mal pieghevole proteine o casuale open reading frame. Abbiamo sviluppato una strategia dove, utilizzando TEM-1 β-lactamase (l'enzima che conferisce resistenza all'ampicillina) su scala genomica, possiamo selezionare collezioni di domini proteina correttamente piegata dalla parte codifica del DNA del genoma qualsiasi intronless. I frammenti di proteine ottenuti da questo approccio, il cosiddetto "domainome", sarà ben espresso e solubile, che li rende adatti per studi strutturali/funzionali.
Clonazione e visualizzando il "domainome" direttamente in un sistema di visualizzazione dei fagi, abbiamo mostrato che è possibile selezionare domini proteici specifici con le proprietà di associazione desiderata (ad es., per altre proteine o anticorpi), fornendo così essenziale informazioni sperimentali per l'identificazione dell'antigene o annotazione genetica.
L'identificazione dei cloni più arricchiti in una popolazione policlonale selezionata può essere realizzato utilizzando tecnologie di sequenziamento di nuova generazione romanzo (NGS). Per queste ragioni, abbiamo introdurre l'analisi di sequenziamento profondo della biblioteca stessa e le uscite di selezione per fornire informazioni complete sulla diversità, abbondanza e mappatura precisa di ogni frammento selezionato. I protocolli qui presentati mostrano i passaggi chiavi per la costruzione della libreria, la caratterizzazione e la convalida.
Introduzione
Qui, descriviamo un metodo di alto-rendimento per la costruzione e la selezione delle biblioteche di domini proteici piegato e solubile da qualsiasi fonte di partenza genica/genomica. L'approccio combina tre diverse tecnologie: phage display, l'uso di un reporter pieghevole e sequenziamento di nuova generazione (NGS) con uno strumento di web specifico per l'analisi dei dati. I metodi possono essere utilizzati in diversi contesti di ricerca basati su proteine, identificazione e annotazione di nuovi domini di proteine/proteine, caratterizzazione delle proprietà strutturali e funzionali di proteine note come pure la definizione di rete di interazione della proteina.
Molte questioni aperte sono ancora presenti nella ricerca di base di proteine e lo sviluppo di metodi per la produzione di proteine ottimale è un bisogno importante per diversi campi di indagine. Ad esempio, nonostante la disponibilità di migliaia di genomi procarioti ed eucarioti1, mappa corrispondente dei proteomi relativi con un'annotazione diretta delle proteine codificate e peptidi è ancora manca per la grande maggioranza degli organismi. Il catalogo dei proteomi completi sta emergendo come un obiettivo impegnativo che richiede uno sforzo enorme in termini di tempo e risorse. Il gold standard per annotazione sperimentale rimane la clonazione di tutti l'Open Reading Frame (ORF) di un genoma, costruendo il così chiamato "ORFeome". Di solito funzione genica viene assegnato in base su omologia di geni correlati di attività nota ma questo approccio è poco accurato a causa della presenza di molte annotazioni errate in riferimento database2,3,4, 5. Inoltre, anche per le proteine che sono state identificate e annotate, ulteriori studi sono richiesti per ottenere la caratterizzazione in termini di abbondanza, modelli di espressione in contesti diversi, incluse le proprietà strutturali e funzionali, nonché reti di interazione.
Inoltre, poiché le proteine sono composte da domini diversi, ognuno di loro che mostrano le caratteristiche specifiche e che contribuiscono in modo diverso alle funzioni della proteina, lo studio e la definizione esatta di questi domini possono consentire un quadro più completo, entrambi presso il singolo gene e a livello del genoma completo. Tutte le informazioni necessarie rendono la ricerca di base di proteine un campo ampio e impegnativo.
In questa prospettiva, un contributo importante potrebbe essere dato dai metodi imparziali e ad alta produttività per la produzione di proteine. Tuttavia, il successo di tali approcci, accanto a notevoli investimenti necessari, si basa sulla capacità di produrre la proteina solubile/stabile costrutti. Questo è un grande fattore di limitazione poiché è stato stimato che solo circa il 30% delle proteine può essere espresso con successo e prodotto a livelli sufficienti per essere sperimentalmente utile6,7,8. Un approccio per superare questa limitazione è basato sull'uso di DNA in modo casuale frammentato per produrre diversi polipeptidi, che insieme offrono sovrapposti rappresentazione di frammento di singoli geni. Solo una piccola percentuale dei frammenti di DNA generati casualmente sono ORFs funzionale mentre la grande maggioranza di loro è non funzionali (per la presenza di codoni di stop all'interno di loro sequenze) o codifica per un naturale (ORF in un frame diverso da quello originale) polipeptidi con nessun significato biologico.
Per affrontare tutti questi problemi, il nostro gruppo ha sviluppato una piattaforma di analisi conforme agli standard della proteina di alto-rendimento espressione e interazione che può essere utilizzata su una scala genomica9,10,11,12. Questa piattaforma integra le seguenti tecniche: 1) un metodo per selezionare raccolte di domini proteina correttamente piegata dalla parte codifica del DNA da qualsiasi organismo; 2) la tecnologia di visualizzazione dei fagi per selezionare i partner delle interazioni; 3) il NGS completamente Interactoma intero sotto studio di caratterizzare e identificare i cloni di interesse; e 4) uno strumento web per l'analisi dei dati per gli utenti senza competenze di programmazione o bioinformatica eseguire analisi Interactoma-Seq in modo semplice e facile da usare.
L'utilizzo di questa piattaforma offre importanti vantaggi rispetto alle strategie alternative di indagine; soprattutto il metodo è completamente imparziale, ad alta produttività e modulare per lo studio che vanno da un singolo gene fino a un intero genoma. Il primo passo della pipeline è la creazione di una libreria dal DNA frammentato in modo casuale in fase di studio, che quindi è profondamente caratterizzata da NGS. Questa libreria viene generata utilizzando un vettore di derivati dal dove vengono clonati geni/frammenti di interesse tra una sequenza di segnale per la secrezione della proteina nello spazio periplasmico (cioè, un leader di Sec) ed il gene di β-lactamase TEM1. La proteina di fusione conferisce resistenza all'ampicillina e la capacità di sopravvivere sotto pressione di ampicillina solo se frammenti clonati sono in-frame con entrambi questi elementi e la proteina di fusione risultante è correttamente piegato10,13 ,14. Tutti i cloni salvato dopo selezione antibiotica, i cosiddetti "cloni filtrata", sono ORFs e, a grande maggioranza di loro (più dell'80%), sono derivati da veri geni9. Inoltre, la potenza di questa strategia sta nei risultati che tutti i cloni di ORF filtrati sono codifica per proteine correttamente piegato/solubile/domini15. Come molti cloni, presenti nella biblioteca e mappatura nella stesso regione/dominio, hanno punti diversi iniziale e finale, in questo modo imparziale, passo singolo identificazione dei frammenti minimi che possono portare a prodotti solubili.
Un ulteriore miglioramento nella tecnologia è dato dall'utilizzo di NGS per caratterizzare la libreria. La combinazione di questa piattaforma e di uno strumento web specifico per l'analisi dei dati dà importanti informazioni imparziali sulle sequenze del nucleotide esatto e sulla posizione di ORFs selezionato sul riferimento del DNA in fase di studio senza la necessità di ulteriori analisi estese o sforzo sperimentale.
Domainome librerie possono essere trasferite in un contesto di selezione e utilizzate come uno strumento universale per eseguire studi funzionali. La proteina di alto-rendimento espressione e interazione analisi piattaforma che abbiamo integrato e che abbiamo chiamato Interactoma-Seq sfrutta la tecnologia di visualizzazione dei fagi trasferimento ORF filtrato in un vettore phagemid e creando un fago-ORF biblioteca. Una volta ri-clonato in un contesto di visualizzazione dei fagi, proteina domini vengono visualizzati sulla superficie delle particelle di M13; in questo modo le librerie domainome possono essere selezionate direttamente per frammenti del gene codifica i domini con specifiche attività enzimatiche o associazione proprietà, permettendo di reti Interactoma profilatura. Questo approccio è stato inizialmente descritto da Zacchi et al. 16 e più tardi usato in diversi altri contesto13,17,18.
Rispetto ad altre tecnologie usate per studiare l'interazione proteina-proteina (tra cui due sistema ibrido del lievito e spettrometria di massa19,20), uno dei principali vantaggi è l'amplificazione del partner associazione che si verifica durante dei fagi visualizzare più cicli di selezione. Questo aumenta la sensibilità della selezione permettendo così l'identificazione dei domini delle proteine bassa associazione abbondanti presenti nella libreria. L'efficienza della selezione eseguita con biblioteca filtrato ORF è ulteriormente incrementata grazie all'assenza di cloni non funzionali. Infine, la tecnologia permette la selezione deve essere eseguita contro proteine e non proteici esche21,22,23,24,25.
Le selezioni dei fagi utilizzando la libreria domainome-fago possono essere eseguite utilizzando anticorpi provenienti dai sieri di pazienti con diverse condizioni patologiche, ad es. malattie di13, cancro o infezione malattie autoimmuni come esca. Questo approccio viene utilizzato per ottenere la cosiddetta "firma di anticorpo" della malattia in fase di studio che permette di identificare e caratterizzare gli antigeni/epitopi specificamente riconosciuti dagli anticorpi dei pazienti allo stesso tempo massiccio. Rispetto ad altri metodi l'uso del fago display permette l'identificazione di epitopi antigenici conformazionali e lineari. L'identificazione di una firma specifica potrebbe potenzialmente avere un impatto importante per la patogenesi di comprensione, nuovo vaccino design, identificazione di nuovi bersagli terapeutici e lo sviluppo di nuovi e specifici strumenti diagnostici e prognostici. Inoltre, quando lo studio si concentra sulle malattie infettive, dei principali vantaggi è che la scoperta delle proteine immunogeniche è indipendente dalla coltivazione di agente patogeno.
Il nostro approccio conferma che i reporter pieghevoli possono essere utilizzati su scala genomica per selezionare il "domainome": un insieme di domini proteici correttamente piegato, bene espresso, solubile dalla parte codifica del DNA e/o cDNA da alcun organismo. Una volta isolato i frammenti di proteine sono utili per molti scopi, fornendo informazioni essenziali sperimentale per l'annotazione del gene anche per quanto riguarda gli studi strutturali, anticorpo epitope mapping, identificazione dell'antigene, ecc. La completezza dei dati ad alta velocità forniti da NGS permette l'analisi di campioni altamente complessi, come ad esempio librerie dei fagi della visualizzazione e possiede il potenziale per eludere la tradizionale raccolta laboriosa e test dei cloni individuali dei fagi salvato.
Allo stesso tempo grazie alle caratteristiche della biblioteca filtrata e per l'estrema sensibilità e la potenza dell'analisi NGS, è possibile identificare il dominio della proteina responsabile di ogni interazione direttamente in una schermata iniziale, senza la necessità di creare librerie aggiuntive per ogni associato della proteina. NGS permette di ottenere una definizione esaustiva dell'intero domainome di qualsiasi origine genica/genomica di partenza e lo strumento di web analisi dati consente l'ottenimento di un'altamente specifica caratterizzazione da un punto di vista qualitativo e quantitativo della domini delle proteine Interactoma.
Protocollo
1. costruzione della biblioteca ORF (Figura 1)
- Preparazione del DNA di inserto
-
Preparazione di frammenti da DNA genomico o sintetico
- Estratto/purificare il DNA utilizzando metodi standard26.
- Frammento di DNA tramite sonicazione. Se utilizzando un sonicatore standard, come un suggerimento generale inizio con 30 impulsi di s al 100% potenza di uscita.
Nota: Gli esperimenti pilota dovrebbero essere fatto con diversa potenza e tempi di sonicazione per impostare le condizioni ottimali per la preparazione di DNA. Dopo ogni test è possibile determinare la dimensione dei frammenti di DNA mediante elettroforesi su gel di agarosio. - Caricare i lisati mediante DNA su gel di agarosio al 1.5%, insieme con un DNA ladder 100 bp. Eseguire un'elettroforesi breve eseguire a 5 V/cm per 15 min e tagliare la parte del gel contenente lo striscio del DNA frammentato.
- Purificare il DNA dell'inserto con un kit di estrazione del gel a base di colonna e misurare la concentrazione utilizzando uno spettrofotometro UV.
Nota: almeno 500 ng di inserti purificate dovrebbero essere ottenute dopo questo passaggio, per essere legato con 1 µ g di vettore digerito, come descritto al punto 1.3. Controllare la qualità della preparazione di frammenti valutando un260nm/A280nm e un260nm/A230nm rapporti poiché bassa qualità del campione influenzerà l'efficienza di legatura. - Trattare fino a 5 μg degli inserti con 1 μL di mix enzima Kit di smussamento rapido, secondo le istruzioni del produttore. Inattivare gli enzimi da riscaldamento a 70 ° C per 10 min campioni possono essere conservati a-20 ° C fino all'utilizzo.
-
Preparazione di frammenti di cDNA
- Estrarre RNA con metodi standard (ad es., usando TRizol o simili reagenti).
- Frammento di mRNA di riscaldamento prima di eseguire la trascrizione inversa. La lunghezza del frammento di DNA finale è controllata da tempo di ebollizione di mRNA e concentrazione casuale dell'iniettore. Ad esempio, riscaldare il campione per 6 min a 95 ° C.
- Preparare cDNA utilizzando primers casuale con qualsiasi kit disponibile seguendo il protocollo del produttore.
- Impoveriscono cDNA delle code di poli-dT tramite l'ibridazione con biotinilati poli-dA per 3 h a 37 ° C e separare il biglie magnetiche streptavidina come descritto da Carninci et al. 13
- Recuperare il materiale non legato e purificare con un kit di purificazione di DNA basata su colonne seguendo le istruzioni del produttore. Misurare la concentrazione utilizzando uno spettrofotometro UV. Vedere la nota nel passaggio 1.1.1.4.
-
Preparazione di frammenti da DNA genomico o sintetico
- Preparazione del filtro vettore
- Digest 5 µ g di clonazione purificata vector pFILTER312 con 10 U di EcoRV degli enzimi di limitazione, protocollo del produttore riportato di seguito.
- Caricare 2 µ l (200 ng) del vettore digerito, insieme a 100 ng del vettore non digerito e 1 marcatore bp a k molecolare, su un gel di agarosio all'1%, per verificare la corretta digestione. Calore inattivare l'enzima di restrizione.
- Aggiungere 1/10 di volume di 10x buffer di fosfatasi e 1 µ l (5 U) di fosfatasi e incubare a 37 ° C per 15 min. calore inattivare per 5 min a 65 ° C.
- Purificare il plasmide digerito mediante estrazione da gel di agarosio e misurare la concentrazione utilizzando uno spettrofotometro UV. Campioni possono essere conservati a-20 ° C fino all'utilizzo.
- La legatura e trasformazione
- Eseguire la legatura come segue: per 1 µ g del plasmide digerito aggiungere 400 ng di inserti fosforilati (plasmide: inserto rapporto molare 1:5), 10 µ l di 10x Buffer per T4 DNA ligasi, 2 µ l di alta concentrazione T4 DNA ligasi in un volume finale di 100 µ l. incubi la reazione a pressione di 16 ° C GHT. Inattivare con il calore a 65 ° C per 10 min.
- Precipitare il prodotto di legatura con l'aggiunta di 1/10 di volume di soluzione di acetato di sodio (3 M, pH 5.2) e 2,5 volumi di etanolo al 100%. Mescolare e congelare a-80 ° C per 20 min.
- Centrifugare a massima velocità per 20 min a 4 ° C. Scartare il surnatante.
- Aggiungere 500 µ l di etanolo 70% freddo per il pellet e centrifugare a massima velocità per 20 min a 4 ° C. Scartare il surnatante.
- Asciugare all'aria il pellet. Risospendere il precipitato DNA in 10 µ l di acqua.
- Eseguire l'elettroporazione delle cellule batteriche.
Nota: L'utilizzo di celle ad alta efficienza (superiore a 5 x 109 transformants per µ g di DNA) è necessario. Si consiglia di utilizzare Escherichia coli DH5αF' (F'/ endA1 hsd17 (rK − mK +) supE44 thi-1recA1 gyrA (Nalr) relA1 (lacZYA-argF) U169 deoR (F80dlacD-(lacZ)M15) prodotto in casa o acquistati da produttori diversi.- Inserire un numero appropriato di microcentrifuga e 0,1 cm-elettroporazione cuvette sul ghiaccio. Aggiungere 1 µ l di soluzione di legatura purificata (in acqua deionizzata) a 25 µ l delle cellule e flick il tubo un paio di volte.
- Trasferire il composto di DNA-cellula in cuvetta fredda tocca sul controsoffitto x 2, pulire acqua sulla parte esterna della cuvetta, posto nell'impulso di modulo e premere elettroporazione.
- Eseguire l'elettroporazione con una macchina di electroporator standard con 25 µF, 200 Ω e 1.8 kV. Costante di tempo deve essere di 4-5 ms.
- Immediatamente aggiungere 1 mL di terreno liquido 2xYT senza alcun antibiotico, trasferire in una provetta 10 mL e lasciar per crescere a 37 ° C, agitando a 220 giri/min per 1 h.
- Piastra trasformato DH5αF' su 15 cm 2xYT piastre di agar completati con 34 cloramfenicolo µ g/mL (pFILTER resistenza) e 25 µ g/mL ampicillina (indicatore selettivo per ORFs) e incubare per una notte a 30 ° C.
- Piastra di diluizioni della libreria su piastre di agar 2xYT 10cm completati con cloramfenicolo + ampicillina e cloramfenicolo solo, per eseguire la titolazione della biblioteca. Incubare per una notte a 30 ° C.
- convalida di biblioteca pFILTER-ORF
- Test di 15-20 colonie da piastre sia cloramfenicolo e cloramfenicolo/ampicillina per stimare la distribuzione delle dimensioni inserto. Scegliere singole colonie con una punta e diluirli separatamente in 100 µ l di terreno di 2xYT senza antibiotici. Utilizzare 0,5 µ l di questa soluzione come modello di DNA per una reazione di PCR, con qualsiasi standard polimerasi di TaqDNA seguendo il protocollo del produttore.
- Eseguire 25 cicli di amplificazione utilizzando una ricottura T di 55 ° C e un tempo di estensione di 40 s a 72 ° C. Sequenze dell'iniettore sono forniti nella Tabella materiali.
- Caricare i prodotti PCR su gel di agarosio 1.5%, insieme a un 100 bp DNA scaletta ed Esegui.
- Collezione Biblioteca pFILTER-ORF
- Raccogliere i batteri dalle piastre 150 mm con l'aggiunta di 3 mL di terreno fresco 2xYT e la loro raccolta con una spatola sterile, mescolare accuratamente, li integrano con 20% glicerolo sterile e conservare a-80 ° C in piccole aliquote.
- Purificare il DNA del plasmide da un'aliquota della biblioteca (prima dell'aggiunta di glicerolo) utilizzando un kit di estrazione basata sulla colonna plasmide, seguendo le istruzioni del produttore.
- Misurare la concentrazione con lo spettrofotometro UV. I campioni possono essere conservati a-20 ° C fino a essere utilizzato per phagemid libreria preparazione e/o caratterizzazione di NGS.
2. subcloning di ORFs filtrato in un vettore Phagemid (Figura 2)
-
Preparazione del ORF filtrata frammenti di DNA
- Impostare la digestione degli enzimi di limitazione di 5 µ g di vettore purificata dal vettore pFILTER-ORF biblioteca aggiungendo 10 U di BssHII e incubando secondo il protocollo del produttore. Inattivare l'enzima e digest con 10 U di NheI.
- Caricare il DNA digerito su gel di agarosio al 1.5%, insieme con un DNA ladder 100 bp. Eseguire un breve elettroforesi eseguito a 5 V/cm per 15 min o appena sufficiente a distinguere la sbavatura dei frammenti asportati e tagliare la parte del gel che li contengono.
- Purificare il DNA dell'inserto con un kit di estrazione di colonna-base in gel e misurare la concentrazione utilizzando uno spettrofotometro UV.
-
Preparazione del phagemid DNA
- Impostare la digestione degli enzimi di limitazione di 5 µ g di purificato pDAN527 per quanto riguarda gli inserti.
- Purificare il plasmide digerito di DNA digerito in esecuzione su un gel di agarosio 0,75% ed estrarre dal gel con un kit di base di colonna.
- Misurare la concentrazione utilizzando uno spettrofotometro UV. Campioni possono essere conservati a-20 ° C fino all'utilizzo.
-
Raccolta, trasformazione e legatura di biblioteca
- Eseguire la legatura e trasformazione come descritto per il vettore di pFILTER.
- Piastra trasformato DH5αF' su 150 mm piastre di agar 2xYT completati con ampicillina di 100 µ g/mL e incubare per una notte a 30 ° C.
- Piastra di diluizioni della libreria su piastre di agar 2xYT 100mm completati con ampicillina 100 di µ g/mL per determinare le dimensioni della libreria.
- Eseguire la convalida della libreria mediante PCR di cloni selezionati in modo casuale come descritto al punto 1.4.
- Raccogliere la libreria phagemid-ORF raccogliendo i batteri da 150 mm piastre, mescolare accuratamente, li integrano con 20% glicerolo sterile e conservare a-80 ° C in piccole aliquote.
- Purificare il DNA del plasmide da un'aliquota della biblioteca utilizzando un kit di estrazione basata sulla colonna plasmide, seguendo le istruzioni del produttore.
- Misurare la concentrazione presso lo spettrofotometro UV. I campioni possono essere conservati a-20 ° C fino a essere utilizzati per la caratterizzazione di NGS.
3. dei fagi libreria preparazione e procedura di selezione
- Produzione dei fagi
- Diluire un'aliquota d'archivio della biblioteca phagemid in 10 mL di brodo liquido 2xYT integrata con ampicillina di 100 µ g/mL al fine di avere un OD600nm = 0,05.
- Crescere la libreria diluita in una beuta sterile 5 - 10 volte più grande rispetto al volume originale, a 37 ° C con agitazione a 220 giri/min fino a quando non raggiunge OD600nm = 0,5.
- Infettare i batteri con fago helper (ad es. M13K07) ad una molteplicità di infezione 20:1. Lasciare a 37 ° C per 45 minuti con agitazione occasionale (ogni 10 min).
- Centrifugare i batteri a 4000 x g per 10 min a temperatura ambiente. Eliminare il surnatante e risospendere il pellet di batteri in 40 mL di brodo liquido 2xYT completato con 100 µ g/mL ampicillina e kanamicina di 50 µ g/mL crescere a 28 ° C con agitazione a 220 giri/min per una notte.
- Il giorno dopo, i batteri della centrifuga a 4000 x g per 20 min a 4 ° C. Raccogliere il surnatante contenente i fagi.
- PEG-precipitazione dei fagi.
- Aggiungere volume 1/5 di un 0,22 µm filtrata PEG/NaCl soluzione (20% w/v PEG 6000, 2,5 M NaCl) per i fagi deselezionati e incubare in ghiaccio per 30-60 min.
Nota: Soluzione è diventato fumoso dopo pochi minuti, che indica una precipitazione di successo dei fagi. La nuvolosità della soluzione aumenterà nel corso del tempo di incubazione. - Centrifugare a 4000 x g per 15 min a 4 ° C. Si formerà una piccola pallina bianca di fagi.
- E risospendere in 1 mL di PBS sterile. Trasferimento a provetta da 1,5 mL e centrifugare a 4 ° C per 10 minuti alla massima velocità per rimuovere i batteri contaminanti. Si formerà una pallina marrone.
- Trasferimento supernatante contenente fagi ad un nuovo tubo. Tenere fagi su ghiaccio per la selezione successiva titolazione e dei fagi.
- Aggiungere volume 1/5 di un 0,22 µm filtrata PEG/NaCl soluzione (20% w/v PEG 6000, 2,5 M NaCl) per i fagi deselezionati e incubare in ghiaccio per 30-60 min.
- Titolazione dei fagi
- Preparare diluizioni seriali della soluzione dei fagi. Mettere 10 µ l della soluzione dei fagi in 990 µ l di PBS per ottenere 10-2 diluizione. Diluire nuovamente questa preparazione per fare 10-4 e da questo ottenere una diluizione di 10-6 .
- Crescere DH5αF' cellule dei batteri in terreno liquido 2xYT a 37 ° C con agitazione fino OD600nm = 0,5 è raggiunto. Trasferire 1 mL di batteri preparati in provetta da 1,5 mL e immediatamente infettare con 1 µ l della diluizione 10-4 dei fagi. Incubare senza agitazione a 37 ° C per 45 min. Ripetere la stessa procedura per la diluizione 10-6 .
- Diluizioni di piastra dei batteri infetti nella piastra 2xYT 100 mm. Mettere la piastra a 30 ° C durante la notte.
- Piastra di 100 µ l di DH5αF non infetti ' su una piastra di agar 2xYT completati con ampicillina 100 di µ g/mL per verificare l'assenza di contaminazione nella preparazione.
- Il giorno dopo il conteggio del numero di colonie e calcolare il titolo dei fagi. Titolo espressa come numero di fagi/mL. Titolo previsto è12-13 fagi/10ml.
- Selezione dei fagi
-
Selezione dei fagi usando come esca purificato anticorpi
- Saturare i fagi diluendo 200 µ l della preparazione dei fagi in un volume uguale di PBS - 4% latte scremato e incubare per 1 h a temperatura ambiente in lenta rotazione. Questo passaggio permette di bloccare dei fagi per il legame non specifico. Trasferire 30 µ l di biglie magnetiche rivestite di proteine-G in una provetta da 1,5 mL.
- Lavare due volte come segue: aggiungere 500 µ l di PBS, incubare su una ruota in rotazione lenta per 2 min a temperatura ambiente, disegnare le perline su un lato del tubo utilizzando un magnete e rimuovere il surnatante.
- Incubare saturi fagi con perline lavati per 30 min a temperatura ambiente con rotazione lenta.
- Disegnare le perline su un lato usando un campo magnetico. Raccogliere il surnatante contenente fagi da utilizzarsi per la fase di selezione.
- Preparare biglie magnetiche durante l'esecuzione del passaggio precedente, coniugando anticorpi purificati. Lavare 30 µ l di biglie magnetiche rivestite di proteine-G come descritto sopra. Diluire 10 µ g di anticorpi purificati in 500 µ l di PBS, aggiungere le perline lavate ed incubare a rotazione lenta a temperatura ambiente per 45 min. lavare due volte con PBS.
Nota: Eseguire due diverse preparazioni di biglie magnetiche: uno con gli anticorpi di interesse e con controllo anticorpi, ad esempio, gli anticorpi purificati da donatori sani. La sequenza degli antigeni selezionati con controllo anticorpi vengono sottratti durante la fase di analisi delle uscite. In alternativa, perline magnetici caricati con anticorpi di controllo può essere utilizzato per eseguire un passaggio di pre-schiarimento di fagi (seguire il protocollo per l'incubazione con perline non-coniugate). - Selezione dei fagi: disegnare perline su un lato del tubo utilizzando un magnete, rimuovere l'ultimo lavaggio, aggiungere fagi e incubare con rotazione lenta a temperatura ambiente per 90 min lavare 5 volte con 500 µ l di PBS-0.1% Tween-20 e 5 volte con PBS.
- Eluire fagi associati, che rappresenta l'output della selezione, mescolando le perline con 1 mL di DH5αF' cellule coltivate a OD600 = 0,5. Incubare i batteri con perline per 45 min a 37 ° C con agitazione occasionale (ogni 10 min). Piastra l'output su una piastra di agar 2xYT 150mm completata con ampicillina di 100 µ g/mL.
- Piastra di 100 µ l di non diluito e di differenti diluizioni dell'output (10-1 a 10-5) per eseguire la titolazione. Il giorno dopo raccogliere i batteri dalle piastre 150 mm con l'aggiunta di 3 mL di terreno fresco 2xYT e la loro raccolta con una spatola sterile, mescolare accuratamente, li integrano con 20% glicerolo sterile e conservare a-80 ° C in piccole aliquote.
- Coltivare un'aliquota nuovamente per eseguire un secondo turno di selezione. Ripetere tutta la procedura panning come descritto sopra, fatta eccezione per le condizioni di lavaggio. In questo caso lavare 10 volte con PBS - 1% Tween-20 (versare la soluzione nel tubo e riversare nuovamente immediatamente). Aggiungere 500 µ l di PBS, quindi incubare su rotazione a temperatura ambiente per 10 min. di eseguire altri 10 lavaggi con PBS. Procedere con il passo di eluizione per quanto riguarda il primo turno di selezione.
- Estrarre il DNA del plasmide da un'aliquota dell'output utilizzando un kit basato su colonna, seguendo le indicazioni del produttore. Memorizzare il plasmide a-20 ° C fino a quando non verrà utilizzato per il sequenziamento profondo.
-
Selezione dei fagi utilizzando come esca proteine ricombinanti
- Saturare i fagi diluendo 200 µ l della preparazione dei fagi in un volume uguale di PBS - 4% latte scremato e incubare per 1 h a temperatura ambiente in lenta rotazione.
- Aggiungere 100 µ l di biglie magnetiche streptavidina. Incubare per 1 h a temperatura ambiente per selezionare streptavidina-associazione fagi. Rimuovere i fagi con streptavidina-associazione disegnando le perline su un lato con un magnete. Prendere surnatante dal passaggio precedente e aggiungere proteine biotinilate (a una concentrazione di 100-550 nM) e incubare su un rotore a temperatura ambiente per 30 min a 1 h.
- Preparare biglie magnetiche: mentre si esegue il passaggio precedente, lavare 100 µ l di biglie streptavidin-magnetiche con PBS e risospendere in PBS 2% latte scremato ed incubare con rotazione a temperatura ambiente per 30 min a 1 h.
- Selezione dei fagi: disegnare perline su un lato del tubo utilizzando un magnete, rimuovere PBS - 2% latte e risospendere perline con mix dei fagi-proteina. Incubare con rotazione lenta a temperatura ambiente per 90 min.
- Disegnare le perline su un lato del tubo utilizzando un magnete, scartare il surnatante e lavate accuratamente con cinque volte con 500 µ l di PBS 0.1% Tween-20. Eseguire eluizione come descritto nella precedente sessione.
-
Selezione dei fagi usando come esca purificato anticorpi
4. fago biblioteca sequenziamento profondo piattaforma (Figura 3)
-
DNA inserisce recupero da pFILTER-ORF-biblioteca, pDAN5-ORF-libreria o le librerie dei fagi selezionati
- Scongelare un'aliquota della biblioteca, quantificarlo mediante spettrofotometro, recuperare DNA inserti dall'amplificazione con primer specifici.
Nota: I primers utilizzati per salvare gli inserti sono legati alla loro estremità 5' di sequenze di adattatori, consentendo così l'indicizzazione successivi delle piscine degli ampliconi ottenuti e il sequenziamento diretto degli inserti di DNA recuperato utilizzando il sequencer. Loro sequenza è nella Tabella materiali. Gli adattatori sono indicati in grassetto, e i primer specifici sono indicati in corsivo. - Utilizzare 2,5 µ l della biblioteca (pFILTER/phagemid/selezionati-fago) come modello di DNA per una reazione di PCR.
- Utilizzare il seguente programma: 95 ° C per 3 min; 25 cicli di 95 ° C per 30 s, 55 ° C per 30 s, 72 ° C per 30 s; 72 ° C per 5 min Hold a 4 ° C.
Nota: A questo punto si consiglia di eseguire 1 µ l di prodotto PCR su un Bioanalyzer o TapeStation per verificare la dimensione degli ampliconi e verifica che siano nell'intervallo corretto.
- Scongelare un'aliquota della biblioteca, quantificarlo mediante spettrofotometro, recuperare DNA inserti dall'amplificazione con primer specifici.
-
Pulizia PCR
- Portare i branelli magnetici (ad es., AMPure) a temperatura ambiente. Trasferire l'intero prodotto PCR dalla provetta PCR in una provetta da 1,5 mL. Vortice i branelli magnetici per 30 s per assicurarsi che le perle sono uniformemente dispersi. Aggiungere 20 µ l di biglie magnetiche in ogni provetta contenente il prodotto PCR, mix pipettando delicatamente. Incubare a temperatura ambiente senza agitare per 5 min.
- Collocare la piastra su un supporto magnetico per 2 min o fino a quando il surnatante è cancellata. Con i prodotti di PCR su supporto magnetico, rimuovere e scartare il surnatante.
- Lavare le perle con etanolo 80% preparati al momento, con i prodotti di PCR su supporto magnetico, come segue: aggiungere 200 µ l di etanolo di 80% preparati al momento per ogni campione bene; Incubare la piastra sul supporto magnetico per 3 s; rimuovere con cautela e scartare il surnatante.
- Eseguire un secondo lavaggio di etanolo, con i prodotti di PCR su supporto magnetico; alla fine del secondo lavaggio estrarre tutto l'etanolo con cautela e consentire le perline per asciugare all'aria per 10 min.
- Rimuovere i prodotti PCR dal supporto magnetico, µ l 17,5 di Tris 10 mM pH 8,5 a ciascuna provetta, pipettare delicatamente su e giù 10 volte, assicurarsi che perline completamente sono risospese. Incubare a temperatura ambiente per 2 min.
- Posizionare il tubo sul supporto magnetico per 2 min o fino a quando non ha eliminato il surnatante, trasferire con cautela 15 µ l del supernatante contenente i prodotti PCR purificati in una nuova provetta da 1,5 mL. Conservare i prodotti di PCR purificati a-15 ° C a-25 ° C per fino ad una settimana se non procedere immediatamente a indice PCR.
-
Indice PCR
Nota: Dopo PCR ripulire, eseguire Indice PCR. Utilizzare il kit di Nextera XT indice; sarà così possibile sequenziare le librerie indicizzate doppie risultante all'interno del multiplex Illumina corre.- Trasferire tutti i 15 μL contenente ogni prodotto purificato in una nuova provetta PCR e impostare la seguente reazione contenente: 15 µ l di prodotto purificato amplicon, 5 µ l di Primer di indice 1 e 5 µ l di Primer di indice 2, 25 µ l di miscela di x PCR 2; volume finale di 50 µ l.
- Eseguire PCR su un termociclatore usando il seguente programma: 95 ° C per 3 min, 8 cicli di 95 ° C per 30 s, 55 ° C per 30 s, 72 ° C per 30 s; 72° C per 5 min, quindi tenere a 4 ° C.
-
PCR pulizia 2
- Seguire lo stesso protocollo descritto nella sezione 4.2 per PCR ripulire con le seguenti modifiche: nel primo passaggio aggiungere 56 µ l di biglie magnetiche per ogni 50 µ l di prodotto di PCR.
- Risospendere le sfere in 27,5 µ l di 10 mM Tris-HCl pH 8,5 nel passaggio finale di purificazione e trasferire 25 µ l in un nuovo tubo (questa è la libreria finale purificata pronta per quantificazione e quindi sequencing).
- Conservare la piastra a-15 ° C a-25 ° C per fino ad una settimana se non procedendo alla quantificazione di biblioteca.
-
Valutazione qualitativa e quantitativa della libreria di sequenziamento
- Dopo la purificazione, eseguire 1 µ l di un 01:10 diluizione della biblioteca finale su un bioanalyzer per verificare le dimensioni e quantificarlo selezionando la regione della traccia finale biblioteca.
- In parallelo è necessario eseguire la quantificazione di libreria di Real Time PCR utilizzando un kit di quantificazione di libreria per il protocollo del produttore.
-
Librerie di sequenziamento
- Piscina le librerie indicizzate duale prodotte in collaborazione con altre biblioteche di sequenziamento indicizzati dual. Sequenza di che questo tipo di libreria generando lungo legge anche, almeno 250 fine accoppiati bp utilizzando sia il Hiseq2500 o gli strumenti di MiSeq per ottenere in prima legge PE 250bp caso e nel secondo caso 300 bp PE legge.
5. analisi dei dati Bioinformatic utilizzando lo strumento di Web Interactoma-Seq
- Analizzare le letture ha provenute dal sequenziamento di pFILTER/phagemid/selezionati-fago biblioteca con la pipeline di analisi dati Interactoma-seq. Lo strumento web è liberamente disponibile al seguente indirizzo: http://interactomeseq.ba.itb.cnr.it/
Risultati
L'approccio di filtraggio è schematizzato nella Figura 1. Può essere utilizzato ogni tipo di DNA intronless. In Figura 1A è rappresentata la prima parte dell'approccio filtraggio: dopo il caricamento su un gel dell'agarosi o un bioanalyzer, una buona frammentazione del DNA di interesse appare come una macchia di frammenti con una distribuzione di lunghezza nella grandezza desiderata di 150-750 bp. Un'immagine rappresentante virtuale gel del DNA frammentato ottenuta è dato. Caricato su gel di agarosio di frammenti sono poi recuperati, fine-riparato e fosforilati e poi clonati in un vettore di pFILTER precedentemente smussata per creare una libreria di frammenti di DNA casuale. Esecuzione di ogni passaggio della procedura di clonazione in condizioni ottimali è necessario per ottenere la libreria di buona qualità con una copertura totale del DNA in fase di studio.
In Figura 1B è rappresentato l'approccio filtraggio: la biblioteca è cresciuta in presenza di cloramfenicolo (pFILTER resistenza) da solo o cloramfenicolo ed ampicillina per selezionare per le colonie di ORF-contenente. Colonie soli avendo un frammento di DNA corrispondente a un ORF producono un β-lactamase funzionale e sopravvivono quando selezione antibiotica è presente. Figura 1 viene illustrato come aumentare pressione selettiva consente la selezione di buona cartella ORFs contro povero cartella quelli. Il risultato atteso è una diminuzione delle dimensioni libreria di circa 20 volte. Più alto numero di cloni superstiti indica insufficiente pressione selettiva.
ORF frammenti possono essere facilmente recuperati dalla biblioteca filtrata per successiva applicazione; per studi di interazione tra la nostra strategia si avvale della tecnologia dei display dei fagi. In Figura 2, sono rappresentati i passaggi principali della costruzione della libreria dei fagi: una biblioteca adeguata è preparata da taglio frammenti filtrati dal vettore pFILTER e ri-clonazione in un plasmide phagemid in fusione con la sequenza che codifica per la Fago capside proteina g3p. Una volta infettati con fago helper, la presenza del vettore nelle cellule di batteri permette la produzione di particelle fagiche visualizzazione ORF-g3p fusione prodotti sulla loro superficie, rendendo così la biblioteca filtrata disponibile per la selezione dei fagi della visualizzazione e ulteriore analisi.
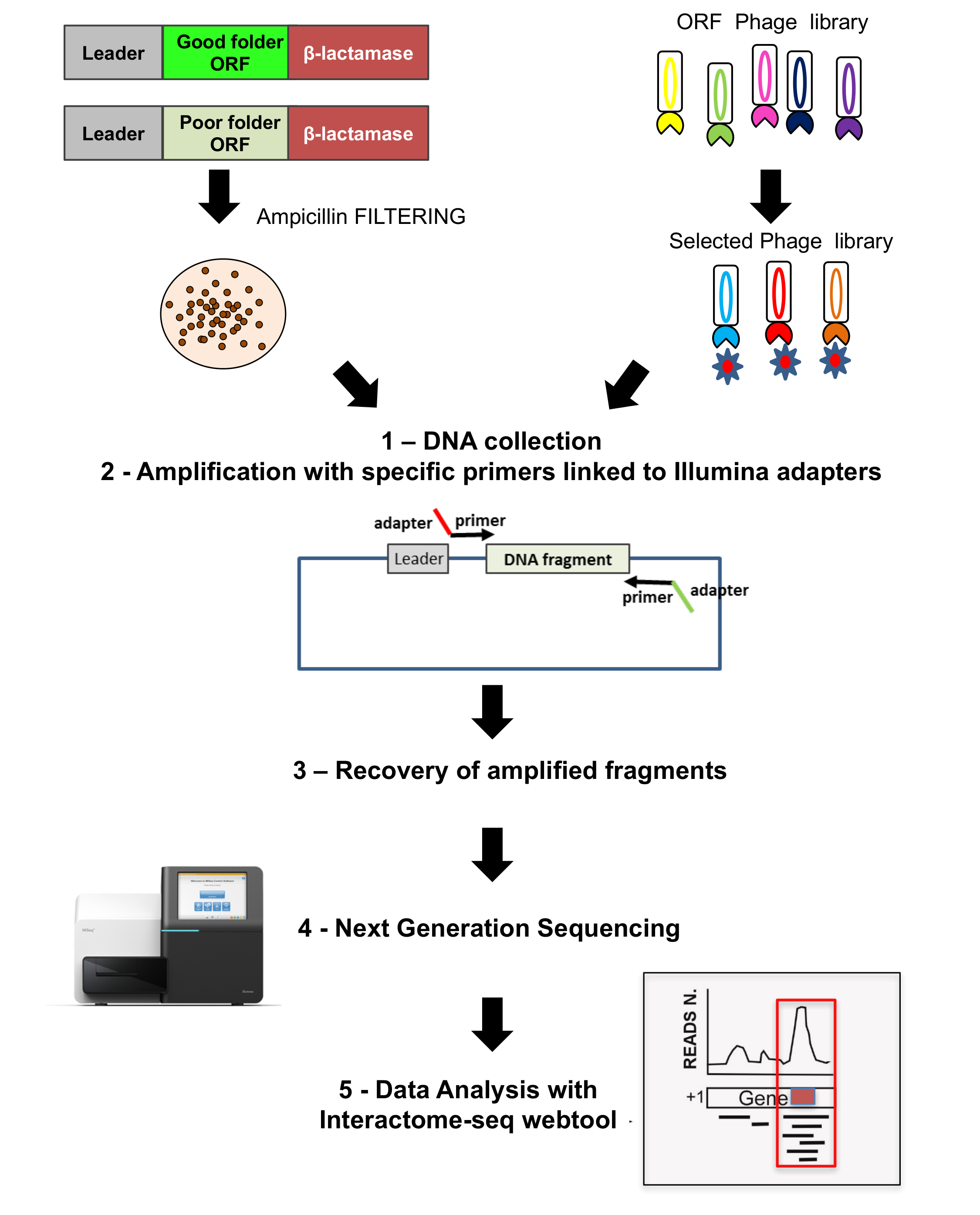
Tutte le librerie vengono analizzate da NGS, come pure le uscite delle selezioni dei fagi, come mostrato nella seconda parte della Figura 3. Frammenti di DNA vengono tratti in salvo dalla crescita di colonie dall'amplificazione di PCR con oligonucleotidi specifici ricottura della spina dorsale del plasmide e portare adattatori specifici per il sequenziamento. NGS è eseguita e letture vengono poi analizzate con lo strumento di web analisi dati Interactoma-Seq.
Nella Figura 4 abbiamo segnalato una rappresentazione schematica della procedura di selezione di una libreria di visualizzazione filtrata dei fagi ORF. La selezione in questo esempio viene eseguita utilizzando anticorpi presenti nei sieri di pazienti affetti da diverse patologie (cioè patologie infettive, patologie autoimmuni, cancro). In questo caso la libreria dei fagi interagisce direttamente con gli anticorpi presenti nei sieri dei pazienti e in questo modo putativi antigeni specifici possono essere arricchiti perché sono riconosciuti dagli anticorpi specifici di malattia. In questo tipo di esperimento, la libreria è anche selezionata solitamente usando i sieri di controllo da pazienti sani al fine di avere un segnale di fondo da utilizzare per le procedure successive di confronto e normalizzazione.
Le selezioni vengono eseguite facendo uso dei sieri provenienti dallo stesso tipo di pazienti solitamente raggruppati in diverse piscine al fine di ridurre la variabilità inter-individuale del titolo dell'anticorpo di sieri. Ogni pool viene utilizzato in modo indipendente per due o tre giri consecutivi della selezione, per arricchire la libreria per immunitario-reattiva cloni specifici per la patologia in esame. Test set anticorpi vengono incubati con fagi biblioteca, immuno-complessi vengono recuperati dalla proteina A rivestito magnetics-perline e fagi associati vengono eluite dalle procedure standard. I cicli di selezione vengono eseguiti con l'aumento di lavaggio e rigore di associazione.
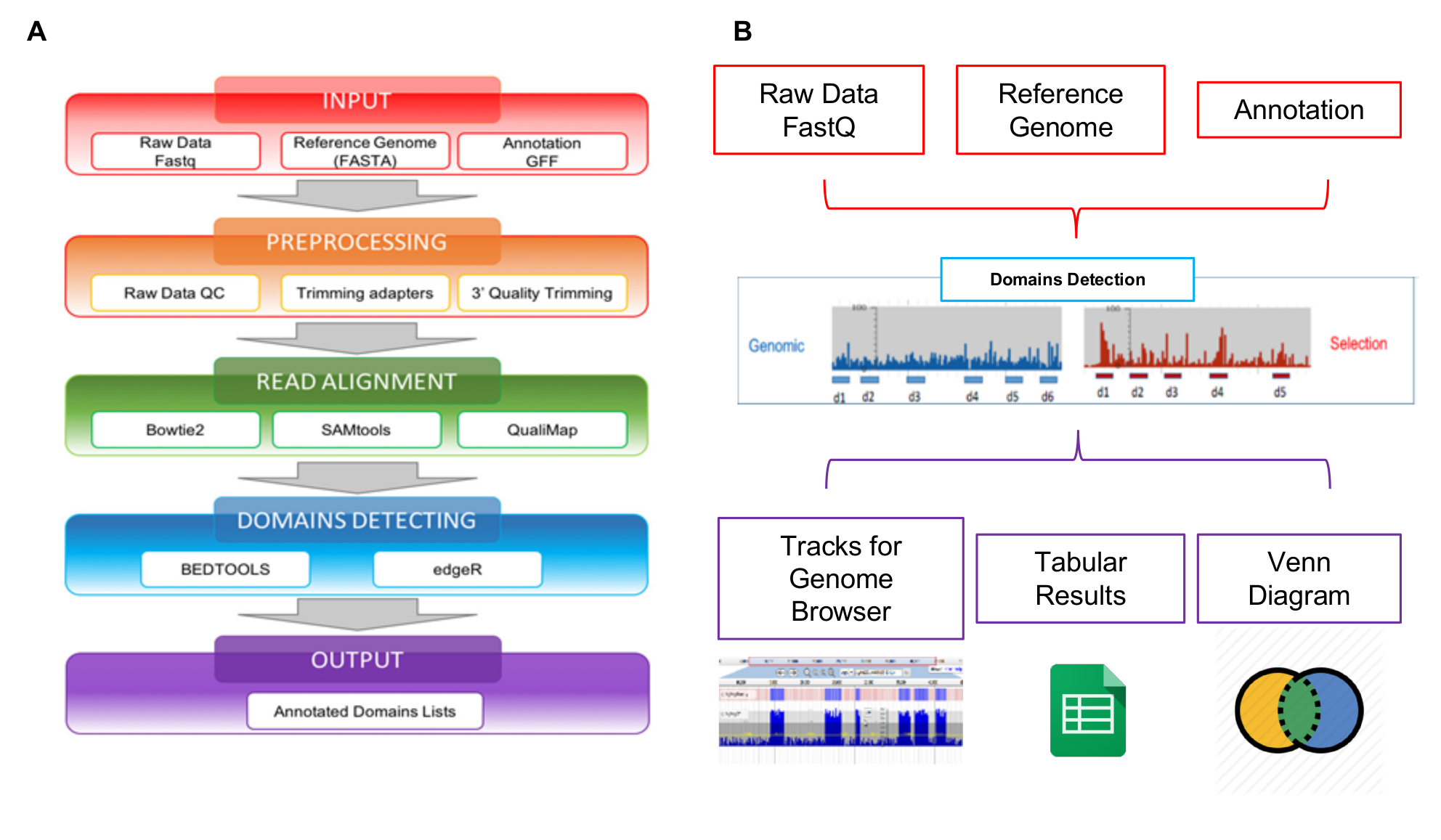
Le letture generate da NGS possono essere analizzate utilizzando lo strumento di web Interactoma-Seq specificamente sviluppato per gestire questo tipo di dati. Flusso di lavoro analisi di dati Interactoma-Seq è composto da quattro fasi sequenziali che, a partire da letture di sequenziamento crudo, genera l'elenco dei domini putativi con annotazioni genomiche (Figura 5A). Nel primo passaggio INPUT (Figura 5A - scatola rossa), Interactoma-Seq controlla se i file di input (crude letture, sequenza del genoma di riferimento, elenco di annotazione) siano correttamente formattati. Nel secondo passaggio pre-elaborazione (Figura 5A - orange box), dati di sequenziamento di bassa qualità vengono prima tagliati utilizzando Cutadapt28 a seconda dei punteggi di qualità e letture con meno di 100 basi di lunghezza vengono scartate. In un passaggio successivo allineamento leggere (Figura 5A - verde casella), il restante letture sono allineate con blastn29 alla sequenza del genoma che permette fino a 5% di mancate corrispondenze. Un file SAM viene generato e legge solo con punteggio di qualità superiore a 30 (Q > 30) vengono elaborati utilizzando SAMtools30 e convertito in un file BAM. Dopo l'allineamento, Interactoma-Seq esegue il rilevamento di domini (Figura 5A - scatola blu), richiamando Bedtools31 per filtrare legge sovrapposte almeno per l'80% della loro lunghezza all'interno di trascrizioni; la copertura, profondità max e valori di messa a fuoco sono quindi calcolati per ogni porzione ORF coperto mappando letture. La copertura rappresenta il numero totale di letture assegnate ad un gene; la profondità è il numero massimo di letture che copre una porzione genica specifica; il focus è un indice ottenuto dal rapporto tra la massima profondità e copertura, e può variare tra 0 e 1. Quando lo stato attivo è superiore a 0,8 e la copertura è superiore la copertura media osservata per tutte le regioni di mapping nel file BAM, la porzione di CD è classificata come un presunto dominio/epitopo. L'ultimo passaggio della pipeline Interactoma-Seq è l'OUTPUT (Figura 5A - contenitore viola), viene generato un elenco di putativi domini in formato tabulare separata. La pipeline Interactoma-Seq è stata inclusa in uno strumento di web per consentire agli utenti senza competenze di programmazione o bioinformatica per eseguire analisi Interactoma-Seq tramite l'interfaccia grafica e di ottenere i loro risultati in un formato semplice e user-friendly. Come mostrato in figura 5B, i risultati di output di un'analisi vengono visualizzati utilizzando JBrowse32 per consentire la visualizzazione e l'esplorazione. Interactoma-Seq genera tracce nel browser del genoma corrispondente putativi domini rilevati e fornisce anche classici diagrammi di Venn per mostrare le intersezioni tra domini putativi comuni arricchite per esempio negli esperimenti di diverse selezioni.

Figura 1: Panoramica schematica dei passaggi principali per la costruzione della biblioteca ORF-filtraggio
A) il DNA da fonte diversa è sonicato e frammentato in frammenti casuali di lunghezza bp 150-750. Frammenti sono recuperati dal gel e clonati come smussato in vector pFILTER; B) filtro passaggio utilizzando β-lactamase come reporter pieghevole. Vettore che contiene frammenti di ORF non vengono selezionate negativamente su ampicillina mentre ORF frammenti clonati permettono le colonie a crescere; C) applicazione di una crescente pressione selettiva (concentrazione di ampicillina nei media di solida crescita da 0 a > 100 μg/mL) consente la selezione di meglio piegati frammenti. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Panoramica schematica dei passaggi principali per la costruzione della libreria dei fagi
A) ORF-filtrata frammenti sono tagliati fuori dal vettore filtrato tramite specifici enzimi di restrizione. Dopo il recupero e purificazione, frammenti sono clonati nel vettore phagemid e trasformati; B) phagemid batterica biblioteca è infetto con fago helper e, dopo una notte crescita, fagi sono PEG-precipitata e raccolti. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Librerie ORF sequenziamento
L'ordinamento viene eseguito su entrambi la libreria selezionata ORF originale, così come sulla libreria phage display; 1) su entrambi i casi vengono recuperati colonie cresciute e DNA Estratto; 2) DNA frammenti vengono recuperati dall'amplificazione utilizzando primers specifici legati agli adattatori per sequenziamento; 3-4) frammenti sono recuperati e profonda sequenziati utilizzando NGS; 5) i dati sono analizzati utilizzando la pipeline Interactoma-Seq. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Schema di selezione biblioteca usando gli anticorpi dei pazienti
Libreria dei fagi è utilizzato per la selezione contro gli anticorpi da sieri dei pazienti. Gli anticorpi sono immobilizzati su biglie magnetiche, viene eseguita l'acquisizione/selezione dei fagi della biblioteca, tre cicli di lavaggi sono effettuati e in seguito fagi selezionati sono recuperati e utilizzati per re-infettare Escherichia coli. Re-infettati Escherichia coli cellule sono placcate in pressione selettiva (ampicillina 100 μg/mL). Frammenti ORF vengono recuperati dall'amplificazione e amplicon piscine sono quindi sequenziate da NGS. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Schema di analisi della libreria
A) rappresentazione del flusso di lavoro di analisi di dati, a partire da file FASTQ raw per le liste di domini con annotazioni finali; B) schematica rappresentazione degli ingressi e delle uscite dello strumento web Interactoma-Seq. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
Discussione
La creazione di una libreria alta qualità altamente diversificata ORFs filtrata è il primo passaggio fondamentale in tutta la procedura poiché interesserà tutti i passaggi successivi della pipeline.
Un'importante caratteristica vantaggiosa del nostro metodo è che qualsiasi fonte di (intronless) del DNA (cDNA, DNA genomic, PCR derivato o DNA sintetico) è adatto per la costruzione della libreria. Il primo parametro che deve essere preso in considerazione è che la lunghezza dei frammenti di DNA clonato nel vettore pFILTER dovrebbe fornire una rappresentazione dell'intera collezione dei domini di un genoma o un transcriptome, il cosiddetto "domainome". Abbiamo dimostrato che domini proteici possono essere clonati con successo, selezionata e infine identificata a partire da frammenti di DNA con una distribuzione lunghezza che va da 150 a 750 bp33,34, e questo è in linea con ciò che è riportato in la letteratura mostrando che la maggior parte dei domini proteici sono di lunghezza aa 100 (con un range da 50 a 200 aa)15.
DNA, il materiale di partenza deve essere frammentato nell'intervallo di dimensioni di scelta e più tardi clonato nel vettore di12 di filtraggio (pFILTER). Durante questi passaggi, potrebbe essere evitato potenziali bias massimizzazione dell'efficienza di tutte la clonazione fasi di reazioni incluse nel protocollo, in particolare frammento fine-riparazione e fosforilazione. La preparazione di vettore è impegnativa e occorre anche, a condizioni ottimali per evitare la degradazione del plasmide e/o contaminazione dal vettore non digerito.
Una volta che la libreria è stata creata, esso dovrebbe essere "filtrata" al fine di mantenere solo ORFs piegati frammenti. Un parametro fondamentale per modulare questo passaggio è la selettiva pressione applicata che possa essere modificata secondo il rigore del filtraggio desiderato. Selezione viene effettuata tramite ampicillina: maggiore è la concentrazione utilizzata, minore è il numero di colonie di batteri trasformati in grado di sopravvivere. Questo riflette la capacità del metodo di filtraggio per selezionare per buono-versus poveri-cartella ORFs34. Questa riduzione del numero di cloni è bilanciata dall'aumento pieghevole proprietà di frammenti selezionati. Di solito, la concentrazione di ampicillina dovrebbe essere sufficiente per ridurre a circa 1/20 il numero di colonie batteriche rispetto a quelli che potrebbero essere ottenuti la libreria il cloramfenicolo solo in crescita.
Convalida di biblioteca è fatto solitamente dall'amplificazione di PCR delle colonie casualmente scelto e loro sequenziamento. L'amplificazione di PCR di alcune colonie è suggerito per avere una rapida stima della qualità della biblioteca: la lunghezza degli inserti dovrebbe essere nell'intervallo previsto di 150-750 bp e diverse colonie dovrebbero presenti inserti con differenti dimensioni buono che indica preparazione di biblioteca in termini di variabilità. Questa strategia convenzionale di screening, quando applicato come unico metodo per la convalida di libreria, non è completa e richiede tempo, che consente l'analisi di un numero limitato di colonie e avendo un'alta probabilità di perdere la maggior parte dei cloni importante. Il nostro approccio si basa sul sequenziamento profondo della libreria, questo fornisce informazioni complete sulla biblioteca diversità e abbondanza e precisa mappatura di ciascuno dei frammenti selezionati.
L'implementazione della tecnologia NGS con l'approccio filtro aumenta la profondità dell'analisi di diversi ordini di grandezza. Recentemente, abbiamo ottimizzato il protocollo per la sequenziazione delle librerie ORF utilizzando la piattaforma Illumina e sviluppato uno strumento web specifico per l'analisi dei dati che rende l'analisi di questi tipi di dati per ogni utente senza qualsiasi bioinformatica competenze di programmazione.
La libreria "per sé" è uno strumento"universale" e può essere sfruttata in diversi contesti di espressione della proteina e/o selezione. Il nostro approccio metodologico si basa sul trasferimento della ORFeome prodotta in un contesto di visualizzazione dei fagi. Frammenti della proteina sono espressi sulla superficie dei fagi e divennero adatti per successiva selezione.
Questo è fatto da salvataggio ORFs filtrata dalla libreria pFILTER di digestione con enzimi di restrizione specifici e ri-duplicarle in un vettore compatibile phagemid permettendo loro fusione con il fago proteina g3p.
Dopo aver creata la libreria phagemid-ORF, può essere utilizzato per la selezione contro obiettivi diversi, come una presunta associazione proteina10 o anticorpi purificati35,36 come descritto qui. Poiché le particelle dei fagi visualizzerà sulla loro superficie filtrata ORFs, ciò si traduce in una procedura di selezione molto più efficace a causa dell'assenza di non visualizzazione cloni che di solito superare esso.
Dopo la selezione della libreria phage display ORF, i cloni di uscita possono essere sequenziati e analizzati con la stessa pipeline. NGS può fornire un completo e statisticamente significativa classifica dei più frequentemente selezionati ORFs e ciò permette l'identificazione delle proteine per lo più interagendo con l'esca utilizzata. Data la presenza di molte versioni diverse di ogni dominio differisce per alcuni aminoacidi, la sovrapposizione tra diversi cloni sequenziati identifica anche il minimo frammento/dominio mostrando proprietà di associazione. Infine, grazie all'abbinamento di genotipo e fenotipo informazioni nella libreria dei fagi, una volta identificati i domini di scelta, la sequenza del DNA può essere facilmente salvata dalla libreria per ulteriori studi, in vitro e in vivo convalida e caratterizzazione.
Divulgazioni
Gli autori non hanno nulla a rivelare.
Riconoscimenti
Questo lavoro è stato supportato da una sovvenzione dal Ministero della pubblica istruzione e Università (2010P3S8BR_002 CP).
Materiali
| Name | Company | Catalog Number | Comments |
| Sonopuls ultrasonic homogenizer | Bandelin | HD2070 | or equivalent |
| GeneRuler 100 bp Plus DNA Ladder | Thermo Scientific | SM0321 | or equivalent |
| GeneRuler 1 kb DNA Ladder | Thermo Fisher Scientific | SM0311 | or equivalent |
| Molecular Biology Agarose | BioRad | 161-3102 | or equivalent |
| Green Gel Plus | Fisher Molecular Biology | FS-GEL01 | or equivalent |
| 6x DNA Loading Dye | Thermo Fisher Scientific | R0611 | or equivalent |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | or equivalent |
| Quick Blunting Kit | New England Biolabs | E1201S | |
| NanoDrop 2000 UV-Vis Spectrophotometer | Thermo Fisher Scientific | ND-2000 | |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | 4368813 | |
| Streptavidin Magnetic Beads | New England Biolabs | S1420S | or equivalent |
| QIAquick PCR purification Kit | Qiagen | 28104 | or equivalent |
| EcoRV | New England Biolabs | R0195L | |
| Antarctic Phosphatase | New England Biolabs | M0289S | |
| T4 DNA Ligase | New England Biolabs | M0202T | |
| Sodium Acetate 3M pH5.2 | general lab supplier | ||
| Ethanol for molecular biology | Sigma-Aldrich | E7023 | or equivalent |
| DH5aF' bacteria cells | Thermo Fisher Scientific | ||
| 0,2 ml tubes | general lab supplier | ||
| 1,5 ml tubes | general lab supplier | ||
| 0,1 cm electroporation cuvettes | Biosigma | 4905020 | |
| Electroporator 2510 | Eppendorf | ||
| 2x YT medium | Sigma-Aldrich | Y1003 | |
| Ampicillin sodium salt | Sigma-Aldrich | A9518 | |
| Chloramphenicol | Sigma-Aldrich | C0378 | |
| DreamTaq DNA Polymerase | Thermo Fisher Scientific | EP0702 | |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | N0447S | |
| 96-well thermal cycler (with heated lid) | general lab supplier | ||
| 150 mm plates | general lab supplier | ||
| 100 mm plates | general lab supplier | ||
| Glycerol | Sigma-Aldrich | G5516 | |
| BssHII | New England Biolabs | R0199L | |
| NheI | New England Biolabs | R0131L | |
| QIAprep Spin Miniprep Kit | Qiagen | 27104 | or equivalent |
| M13KO7 Helper Phage | GE Healthcare Life Sciences | 27-1524-01 | |
| Kanamycin sulfate from Streptomyces kanamyceticus | Sigma-Aldrich | K1377 | |
| Polyethylene glycol (PEG) | Sigma-Aldrich | P5413 | |
| Sodium Cloride (NaCl) | Sigma-Aldrich | S3014 | |
| PBS | general lab supplier | ||
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher Scientific | 10003D | or equivalent |
| MagnaRack Magnetic Separation Rack | Thermo Fisher Scientific | CS15000 | or equivalent |
| Tween 20 | Sigma-Aldrich | P1379 | |
| Nonfat dried milk powder | EuroClone | EMR180500 | |
| KAPA HiFi HotStart ReadyMix | Kapa Biosystems, Fisher Scientific | 7958935001 | |
| AMPure XP beads | Agencourt, Beckman Coulter | A63881 | |
| Nextera XT dual Index Primers | Illumina | FC-131-2001 or FC-131-2002 or FC-131-2003 or FC-131-2004 | |
| MiSeq or Hiseq2500 | Illumina | ||
| Spectrophotomer | Nanodrop | ||
| Agilent Bioanalyzer or TapeStation | Agilent | ||
| Forward PCR primer | general lab supplier | 5’ TACCTATTGCCTACGGCA GCCGCTGGATTGTTATTACTC 3’ | |
| Reverse PCR primer | general lab supplier | 5’ TGGTGATGGTGAGTACTA TCCAGGCCCAGCAGTGGGTTTG 3’ | |
| Forward primer for NGS | general lab supplier | 5’ TCGTCGGCAGCGTCAGA TGTGTATAAGAGACAGGCA GCAAGCGGCGCGCATGC 3’; | |
| Reverse primer for NGS | general lab supplier | 5’ GTCTCGTGGGCTCGGAGA TGTGTATAAGAGACAGGGG ATTGGTTTGCCGCTAGC 3’; |
Riferimenti
- Loman, N. J., Pallen, M. J. Twenty years of bacterial genome sequencing. Nat Rev Microbiol. 13 (12), 787-794 (2015).
- Jones, C. E., Brown, A. L., Baumann, U. Estimating the annotation error rate of curated GO database sequence annotations. BMC Bioinformatics. 8 (1), 170 (2007).
- Andorf, C., Dobbs, D., Honavar, V. Exploring inconsistencies in genome-wide protein function annotations: a machine learning approach. BMC Bioinformatics. 8 (1), 284 (2007).
- Wong, W. -. C., Maurer-Stroh, S., Eisenhaber, F. More Than 1,001 Problems with Protein Domain Databases: Transmembrane Regions, Signal Peptides and the Issue of Sequence Homology. PLoS Comput Biol. 6 (7), e1000867 (2010).
- Bioinformatics, B., et al. Identification and correction of abnormal, incomplete and mispredicted proteins in public databases. BMC Bioinformatics. 9 (9), (2008).
- Phizicky, E., Bastiaens, P. I. H., Zhu, H., Snyder, M., Fields, S. Protein analysis on a proteomic scale. Nature. 422 (6928), 208-215 (2003).
- DiDonato, M., Deacon, A. M., Klock, H. E., McMullan, D., Lesley, S. A. A scaleable and integrated crystallization pipeline applied to mining the Thermotoga maritima proteome. J Struct Funct Genomics. 5 (1-2), 133-146 (2004).
- Nordlund, P., et al. Protein production and purification. Nat Methods. 5 (2), 135-146 (2008).
- Zacchi, P., Sblattero, D., Florian, F., Marzari, R., Bradbury, A. R. M. Selecting open reading frames from DNA. Genome Res. 13 (5), 980-990 (2003).
- Di Niro, R., et al. Rapid interactome profiling by massive sequencing. Nucleic Acids Res. 38 (9), e110 (2010).
- Gourlay, L. J., et al. Selecting soluble/foldable protein domains through single-gene or genomic ORF filtering: Structure of the head domain of Burkholderia pseudomallei antigen BPSL2063. Acta Crystallogr Sect D Biol Crystallogr. 71 (Pt 11), 2227-2235 (2015).
- D'Angelo, S., et al. Filtering "genic" open reading frames from genomic DNA samples for advanced annotation. BMC Genomics. 12 (Suppl 1), S5 (2011).
- D'Angelo, S., et al. Profiling celiac disease antibody repertoire. Clin Immunol. 148 (1), 99-109 (2013).
- Robinson, M. D., McCarthy, D. J., Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26 (1), 139-140 (2009).
- Heger, A., Holm, L. Exhaustive enumeration of protein domain families. J Mol Biol. 328 (3), 749-767 (2003).
- Zacchi, P., Sblattero, D., Florian, F., Marzari, R., Bradbury, A. R. M. Selecting open reading frames from DNA. Genome Res. 13 (5), 980-990 (2003).
- Faix, P. H., Burg, M. A., Gonzales, M., Ravey, E. P., Baird, A., Larocca, D. Phage display of cDNA libraries: Enrichment of cDNA expression using open reading frame selection. Biotechniques. 36 (6), 1018-1029 (2004).
- Patrucco, L., et al. Identification of novel proteins binding the AU-rich element of α-prothymosin mRNA through the selection of open reading frames (RIDome). RNA Biol. 12 (12), 1289-1300 (2015).
- Collins, M. O., Choudhary, J. S. Mapping multiprotein complexes by affinity purification and mass spectrometry. Curr Opin Biotechnol. 19 (4), 324-330 (2008).
- Suter, B., Kittanakom, S., Stagljar, I. Two-hybrid technologies in proteomics research. Curr Opin Biotechnol. 19 (4), 316-323 (2008).
- Nakai, Y., Nomura, Y., Sato, T., Shiratsuchi, A., Nakanishi, Y. Isolation of a Drosophila gene coding for a protein containing a novel phosphatidylserine-binding motif. J Biochem. 137 (5), 593-599 (2005).
- Deng, S. J., et al. Selection of antibody single-chain variable fragments with improved carbohydrate binding by phage display. J Biol Chem. 269 (13), 9533-9538 (1994).
- Danner, S., Belasco, J. G. T7 phage display: A novel genetic selection system for cloning RNA-binding proteins from cDNA libraries. Proc Natl Acad Sci. 98 (23), 12954-12959 (2001).
- Gargir, A., Ofek, I., Meron-Sudai, S., Tanamy, M. G., Kabouridis, P. S., Nissim, A. Single chain antibodies specific for fatty acids derived from a semi-synthetic phage display library. Biochim Biophys Acta - Gen Subj. 1569 (1-3), 167-173 (2002).
- Patrucco, L., et al. Identification of novel proteins binding the AU-rich element of α-prothymosin mRNA through the selection of open reading frames (RIDome). RNA Biol. 12 (12), 1289-1300 (2015).
- Ausubel, F. M., et al. Current Protocols in Molecular Biology. Mol Biol. 1 (2), 146 (2003).
- Sblattero, D., Bradbury, A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat Biotechnol. 18, 75-80 (2000).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 17 (1), 10 (2011).
- Camacho, C., et al. BLAST+: architecture and applications. BMC Bioinformatics. 10 (1), 421 (2009).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Quinlan, A. R. BEDTools: The Swiss-Army tool for genome feature analysis. Curr Protoc Bioinforma. , (2014).
- Skinner, M. E., Uzilov, A. V., Stein, L. D., Mungall, C. J., Holmes, I. H. JBrowse: A next-generation genome browser. Genome Res. 19 (9), 1630-1638 (2009).
- Gourlay, L. J., et al. Selecting soluble/foldable protein domains through single-gene or genomic ORF filtering: Structure of the head domain of Burkholderia pseudomallei antigen BPSL2063. Acta Crystallogr Sect D Biol Crystallogr. 71, 2227-2235 (2015).
- D'Angelo, S., et al. Filtering "genic" open reading frames from genomic DNA samples for advanced annotation. BMC Genomics. 12 (Suppl 1), S5 (2011).
- Di Niro, R., et al. Characterizing monoclonal antibody epitopes by filtered gene fragment phage display. Biochem J. 388 (Pt 3), 889-894 (2005).
- D'Angelo, S., et al. Profiling celiac disease antibody repertoire. Clin Immunol. 148 (1), 99-109 (2013).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati