
Method Article
Interactoma-Seq: Un protocolo para la biblioteca de Domainome construcción, validación y selección de Phage Display y la siguiente secuencia de generación
En este artículo
Resumen
Los protocolos descritos permiten la construcción, caracterización y selección (contra el blanco de la opción) de una biblioteca de "domainome" de cualquier fuente de ADN. Esto se logra mediante una tubería de investigación que combina diferentes tecnologías: exhibición phage, un reportero plegable y próxima generación la secuencia con una herramienta web de análisis de datos.
Resumen
Plegable de reporteros son proteínas con fenotipos fácilmente identificables, tales como resistencia a los antibióticos, cuyo plegamiento y función está comprometido cuando fusionado al mal plegamiento de proteínas o marcos de lectura abierto al azar. Hemos desarrollado una estrategia que, mediante el uso de β-lactamasa TEM-1 (la enzima que confiere resistencia a la ampicilina) en una escala genomic, podemos seleccionar colecciones de dominios de la proteína plegada correctamente desde la parte de codificación de la DNA de cualquier genoma intronless. Los fragmentos de proteína obtenidos por este planteamiento, el así llamado "domainome", será bien expresado y soluble, haciéndolas adecuadas para estudios estructurales/funcionales.
Por clonación y mostrando la "domainome" directamente en un sistema de visualización de fagos, hemos demostrado que es posible seleccionar los dominios de la proteína específica con las propiedades de enlace deseado (por ejemplo, a otras proteínas o anticuerpos), proporcionando así esencial información experimental para la identificación de genes anotación o antígeno.
La identificación de los clones más enriquecidos en una población policlonal seleccionada puede lograrse mediante el uso de tecnologías de secuenciación de próxima generación novela (NGS). Por estas razones, presentamos análisis de secuenciación profunda de la propia biblioteca y las salidas de selección para proveer información completa sobre la diversidad, abundancia y asignación precisa de cada uno de lo fragmento seleccionado. Los protocolos presentados aquí muestran los pasos para la construcción de la biblioteca, caracterización y validación.
Introducción
Aquí, describimos un método de alto rendimiento para la construcción y selección de las bibliotecas de los dominios de la proteína soluble y doblado de cualquier fuente partida genic/genómica. El enfoque combina tres tecnologías diferentes: exhibición phage, el uso de un reportero plegable y siguiente generación de secuenciación (NGS) con una herramienta específica para análisis de datos. Los métodos se pueden utilizar en muchos contextos diferentes de investigación basados en proteínas, para la identificación y anotación de nuevos dominios de las proteínas/proteína, caracterización de propiedades estructurales y funcionales de las proteínas conocidas como definición de red de interacción de la proteína.
Muchas preguntas abiertas todavía están presentes en la investigación basada en la proteína y el desarrollo de métodos para la producción de proteína óptima es una necesidad importante de varios campos de investigación. Por ejemplo, a pesar de la disponibilidad de miles de genomas procariotas y eucariotas1correspondiente mapa de proteomas relativa con una anotación directa de los péptidos y proteínas codificadas es todavía faltando para la gran mayoría de los organismos. El catálogo de proteomas completos está emergiendo como una meta difícil que requiere un gran esfuerzo en términos de tiempo y recursos. El estándar de oro para anotación experimental sigue siendo la clonación de todos los abiertos marcos de lectura (ORFs) de un genoma, construyendo el llamado "ORFeome". Funciones de los genes es asignado generalmente basada en homología con genes relacionados de actividad conocida, pero esta aproximación es poco exacta debido a la presencia de muchas anotaciones incorrectas en referencia bases de datos2,3,4, 5. Por otra parte, incluso para las proteínas que se han identificado y anotado, estudios adicionales son necesarios para lograr la caracterización en términos de abundancia, patrones de expresión en diversos contextos, incluyendo propiedades estructurales y funcionales, así como redes de interacción.
Además, puesto que las proteínas se componen de diferentes dominios, cada uno de ellos que muestran características específicas y diferentemente que contribuyen a las funciones de la proteína, el estudio y la definición exacta de estos dominios pueden permitir una imagen más amplia, tanto en el solo gen y a nivel de genoma completo. Toda esta información necesaria hace investigación basada en la proteína un campo amplio y desafiante.
En esta perspectiva, podría darse una importante contribución por métodos imparciales y alto rendimiento para la producción de proteína. Sin embargo, el éxito de estos enfoques, al lado de la inversión considerable, se basa en la capacidad de producir construcciones de la proteína soluble/estable. Esto es una gran limitación de factor ya que se estima que sólo alrededor del 30% de proteínas puede correctamente expresado y producido en suficientes niveles experimentalmente útil6,7,8. Un enfoque para superar esta limitación se basa en el uso de ADN fragmentado al azar para producir polipéptidos diferentes, que juntos proporcionan representación superpuestos de fragmento de genes individuales. Sólo un pequeño porcentaje de los fragmentos de ADN generados al azar son ORFs funcionales mientras que la gran mayoría de ellos es no-funcionales (debido a la presencia de codones de parada dentro de sus secuencias) o codifica para no natural (ORF en un marco que no sea la original) polipéptidos con ningún significado biológico.
Para abordar todas estas cuestiones, nuestro grupo ha desarrollado una proteína de alto rendimiento expresión e interacción plataforma de análisis que puede utilizarse en una escala genomic9,10,11,12. Esta plataforma integra las siguientes técnicas: 1) un método para seleccionar colecciones de dominios de la proteína correctamente doblada de la parte de codificación de ADN de cualquier organismo; 2) la tecnología phage para la selección de socios de las interacciones; 3) la NGS completamente caracterizar el interactoma conjunto objeto de estudio e identificar los clones de interés; y 4) una herramienta web para el análisis de datos para usuarios sin conocimientos de programación ni bioinformática realizar análisis de Seq interactoma en forma sencilla y fácil de usar.
El uso de esta plataforma ofrece ventajas importantes sobre estrategias alternativas de investigación; sobre todo el método es completamente imparcial, alto rendimiento y modular para el estudio de un solo gen hasta un genoma entero. El primer paso de la tubería es la creación de una biblioteca de ADN fragmentado aleatoriamente bajo estudio, que luego se caracteriza profundamente por NGS. Esta biblioteca se genera usando un vector de ingeniería donde se clonan genes/fragmentos de interés entre una secuencia de la señal para la secreción de proteínas en el espacio periplasmic (es decir, un líder de la Sec) y el gen de β-lactamasa TEM1. La proteína de fusión confiere resistencia a la ampicilina y la capacidad de sobrevivir bajo la presión de la ampicilina solamente si fragmentos clonados en marco con estos elementos y la proteína de fusión resultante es correctamente doblada10,13 ,14. Todos los clones rescataron después de la selección de antibióticos, los llamados "clones de filtrado", son ORFs y, una gran mayoría de ellos (más del 80%), se derivan de genes reales9. Por otra parte, el poder de esta estrategia radica en los resultados que todos los clones ORF filtrado son codificación de proteínas correctamente doblado/soluble/dominios15. Como muchos clones, presentes en la biblioteca y la cartografía en el misma región/dominio, tienen diferentes empezando y terminando puntos, esto permite identificar imparcial, solo paso los fragmentos mínimos que pueden resultar en productos solubles.
Otra mejora en la tecnología se da por el uso de NGS para caracterizar a la biblioteca. La combinación de esta plataforma y de una herramienta de web específica para el análisis de los datos da importante información imparcial sobre las secuencias de nucleótido exacta y la ubicación de las ORFs en la referencia de ADN objeto de estudio sin necesidad de análisis más extensos o esfuerzo experimental.
Bibliotecas de Domainome pueden ser transferidas en un contexto de selección y utilizadas como un instrumento universal para llevar a cabo estudios funcionales. El alto rendimiento proteína expresión e interacción plataforma de análisis que integramos y que llamamos interactoma Seq se aprovecha de la tecnología de exhibición phage por transferir el ORF filtrado en un vector de phagemid y la creación de una fago-ORF Biblioteca. Una vez volver a clonar en un contexto de exhibición phage, proteína dominios aparecen en la superficie de las partículas de M13; de esta manera domainome las bibliotecas pueden seleccionarse directamente para fragmentos génicos codifican dominios con actividades enzimáticas específicas o enlace propiedades, permitiendo redes interactoma perfiles. Este enfoque fue inicialmente descrito por Zacchi et al. 16 y utilizado más adelante en varios otros contexto13,17,18.
En comparación con otras tecnologías utilizadas para el estudio de la interacción de proteínas (incluyendo sistema de híbrido dos levaduras y espectrometría de masas19,20), una ventaja importante es la amplificación del socio de enlace que se produce durante el fago Mostrar múltiples rondas de selección. Esto aumenta la sensibilidad de selección permitiendo así la identificación de dominios de bajo abundantes proteínas presentes en la biblioteca. La eficiencia de la selección realizada con filtrado de ORF biblioteca es incrementada debido a la ausencia de clones no funcional. Por último, la tecnología permite la selección a realizar contra la proteína y la proteína no cebos21,22,23,24,25.
Selección de fagos usando la biblioteca domainome-phage se puede realizar usando los anticuerpos procedentes de sueros de pacientes con diferentes condiciones patológicas, por ejemplo enfermedades autoinmunes13, cáncer o infección enfermedades como cebo. Este enfoque se utiliza para obtener la llamada "firma del anticuerpo" de la enfermedad bajo estudio permitiendo masivamente identificar y caracterizar los antígenos/epitopos específicamente reconocidos por los anticuerpos de los pacientes al mismo tiempo. En comparación con otros métodos el uso de phage display permite la identificación de epítopos antigénicos lineal y conformacional. La identificación de una firma específica podría potencialmente tener un impacto importante para entender patogenesia, nuevo diseño de la vacuna, identificación de nuevas dianas terapéuticas y el desarrollo de herramientas nuevas y específicas de diagnósticos y pronósticos. Por otra parte, cuando el estudio se centra en las enfermedades infecciosas, una ventaja importante es que el descubrimiento de proteínas inmunogénicas es independiente de cultivo del patógeno.
Nuestro enfoque confirma que los reporteros plegables se pueden utilizar en una escala genomic para seleccionar "domainome": una colección de dominios correctamente doblada, bien expresado, solubilidad de la proteína de la parte de codificación de la DNA o cDNA de cualquier organismo. Una vez aislados los fragmentos de proteínas son útiles para muchos propósitos, que proporciona información experimental esencial para anotación del gene así como por los estudios estructurales, Mapeo epitopo de anticuerpo, identificación de antígeno, etcetera. La integridad de datos de alto rendimiento NGS permite el análisis de muestras altamente complejos, tales como bibliotecas de exhibición phage y tiene el potencial para eludir la tradicional cosecha laboriosa y prueba de clones individuales phage rescatado.
Al mismo tiempo gracias a las características de la biblioteca de filtrado y a la extrema sensibilidad y potencia de los análisis NGS, es posible identificar el dominio de la proteína responsable de cada interacción directamente en una pantalla inicial, sin necesidad de crear bibliotecas adicionales para cada destino proteína. NGS permite para obtener una definición completa de la domainome entera de cualquier fuente partida genic/genómica y la herramienta web de análisis de datos permite la obtención de una caracterización muy específica desde un punto de vista cualitativo y cuantitativo de la Dominios del interactoma proteínas.
Protocolo
1. construcción de la biblioteca de la ORF (figura 1)
- Preparación del inserto de ADN
-
Preparación de fragmentos de ADN genómico o sintético
- Extracto/purificar DNA usando métodos estándar26.
- Fragmento de ADN por sonicación. Si utilizando un sonicador estándar, como un comienzo de sugerencia general con 30 pulsos de s al 100% de potencia.
Nota: Los experimentos piloto deben hacerse con diversa energía y tiempos de sonicación para establecer las condiciones óptimas para la preparación de ADN. Después de cada prueba determinar el tamaño de los fragmentos de ADN por electroforesis en gel de agarosa. - Carga de la sonicación DNA en gel de agarosa al 1.5%, junto con una escalera de DNA 100 bp. Realizar una electroforesis corto a 5 V/cm durante 15 minutos y cortar la porción del gel que contiene el borrón de transferencia del ADN fragmentado.
- Purificar el ADN inserto con un kit de extracción de gel base de columna y medir la concentración utilizando un espectrofotómetro UV.
Nota: al menos 500 ng de insertos purificadas debe obtenerse después de este paso, al estar ligada con 1 μg de vector digerido, como se describe en el paso 1.3. Comprobar la calidad de la preparación de fragmentos evaluando un260nm/A280nm y un /A260nm230nm proporciones ya que la baja calidad de la muestra afecta la eficacia de la ligadura. - Tratar hasta 5 μg de los insertos con 1 μL de la mezcla de enzima Kit rápido de embotar, según instrucciones del fabricante. Inactivar enzimas calentando a 70 ° C por 10 minutos las muestras pueden almacenarse a-20 ° C hasta su uso.
-
Preparación de fragmentos de cDNA
- Extracto de RNA con métodos estándar (por ejemplo, usando TRizol o reactivos similares).
- Fragmento de ARNm por calentamiento antes de realizar la transcripción inversa. La longitud final del fragmento de ADN es controlada por el tiempo de ebullición de mRNA y la concentración de cartilla al azar. Por ejemplo, calentar la muestra durante 6 min a 95 ° C.
- Preparar el cDNA usando las cartillas al azar con cualquier kit disponible siguiendo el protocolo del fabricante.
- Agotar los cDNA de colas de poli-dT por hibridación con biotinilado poli-dA por 3 h a 37 ° C y separar en granos magnéticos estreptavidina según lo descrito por Carninci et al. 13
- Recuperar el material no Unido y purificar con un kit de purificación de DNA basado en columna siguiendo las instrucciones del fabricante. Medir la concentración utilizando un espectrofotómetro UV. Ver nota en paso 1.1.1.4.
-
Preparación de fragmentos de ADN genómico o sintético
- Preparación del vector filtrado
- Digest 5 μg de clonación purificada vector pFILTER312 con 10 U de EcoRV enzima de restricción, protocolo de siguiente del fabricante.
- Carga 2 μl (200 ng) del vector digerido, junto con 100 ng de vector no digerido y 1 k bp molecular marcador, en un gel de agarosa al 1%, para verificar la correcta digestión. Inactive por calor el enzima de restricción.
- Volumen 1/10 de 10 x buffer de fosfatasa y 1 μl (5 U) de fosfatasa e incubar a 37 ° C para inactivar los 15 minutos calor durante 5 minutos a 65 ° C.
- Purificar el plásmido digerido por la extracción de gel de agarosa y medir la concentración utilizando un espectrofotómetro UV. Las muestras pueden almacenarse a-20 ° C hasta su uso.
- Ligadura y transformación
- Realizar ligadura como sigue: 1 μg de plásmido digerido añadir 400 ng de insertos fosforiladas (plásmido: insertar relación molar 1:5), 10 μl de 10 x tampón para T4 ADN ligasa, 2 μl de alta concentración T4 ADN ligasa en un volumen final de 100 μl. Incubar la reacción en overni de 16 ° C GHT. Inactive por calor a 65 ° C durante 10 minutos.
- Precipitar el producto de la ligadura mediante la adición de 1/10 volumen de solución de acetato de sodio (3 M, pH 5.2) y 2,5 volúmenes de etanol al 100%. De la mezcla y congelar a-80 ° C durante 20 minutos.
- Centrifugar a velocidad máxima durante 20 min a 4 ° C. Deseche el sobrenadante.
- Añadir 500 μl de etanol frío al 70% para el pellet y centrifugar a máxima velocidad por 20 min a 4 ° C. Deseche el sobrenadante.
- Secar el pellet. Resuspender el ADN precipitado en 10 μl de agua.
- Realizar electroporación de la célula bacteriana.
Nota: El uso de células de alta eficiencia (por encima de 5 x 109 transformantes por μg de ADN) se requiere. Le sugerimos que use Escherichia coli DH5αF' (F'/ endA1 hsd17 (rK − mK +) supE44 thi-1recA1 gyrA (Nalr) relA1 (lacZYA argF) U169 deoR (F80dlacD-(lacZ)M15) producido en casa o comprados de varios fabricantes.- Coloque el número apropiado de 0,1 cm-electroporación cubetas y tubos de microcentrífuga en hielo. Añadir 1 μl de la solución purificada de la ligadura (en DI agua) a 25 μl de las células y el tubo de la película un par de veces.
- Transferir la mezcla de ADN de la célula a la cubeta fría Pulse sobre encimera 2 x, limpie el agua desde el exterior de la cubeta, coloque en el pulso de módulo y presione de electroporación.
- Realizar electroporación con una máquina estándar electroporator usando 25 μF, Ω 200 y 1,8 kV. Constante de tiempo debe ser ms 4-5.
- Añadir 1 mL de medio 2xYT líquido sin ningún antibiótico, transferir a un tubo de 10 mL y permitir crecer a 37 ° C, agitando a 220 rpm durante 1 h.
- Transformado de placa DH5αF' 2xYT de 15 cm las placas de agar suplementado con 34 μg/mL cloranfenicol (pFILTER resistencia) y ampicilina (marcador selectivo de ORFs) de 25 μg/mL e incubar durante una noche a 30 ° C.
- Diluciones de la placa de la biblioteca en placas de agar de 2xYT 10 cm complementado con cloranfenicol + ampicilina y cloranfenicol, para realizar la valoración de la biblioteca. Incubar durante una noche a 30 ° C.
- validación de biblioteca pFILTER ORF
- Prueba de 15 a 20 colonias de las placas de cloranfenicol y ampicilina/cloramfenicol para estimar la distribución de tamaño de inserto. Seleccionar colonias solo con una punta y diluirles por separado en 100 μl de medio 2xYT sin antibióticos. Utilizar 0,5 μl de esta solución como plantilla de la DNA para una reacción de PCR, con cualquier estándar polimerasa TaqDNA siguiendo el protocolo del fabricante.
- Realizar 25 ciclos de amplificación utilizando un recocido T de 55 ° C y un tiempo de extensión de 40 s a 72 ° C. Secuencias de la cartilla se encuentran en la Tabla de materiales.
- Los productos PCR en gel de agarosa al 1.5%, junto con una 100 bp ADN escalera y gestión de la carga.
- colección de la biblioteca pFILTER ORF
- Recoger las bacterias de las placas de 150 mm añadiendo 3 mL de medio 2xYT fresco y cosecharlos con un raspador estéril, mezclar bien, complementar con el 20% de glicerol estéril y almacenar a-80 ° C en alícuotas pequeñas.
- Purificar el DNA plasmídico de una alícuota de la biblioteca (antes de la adición de glicerol) con un kit de extracción de plásmidos basado en la columna, siguiendo las instrucciones del fabricante.
- Medir la concentración con el espectrofotómetro de UV. Las muestras pueden conservarse a-20 ° C hasta ser utilizado para preparación de biblioteca phagemid o caracterización de NGS.
2. subcloning de ORFs filtradas en un Vector de Phagemid (figura 2)
-
Preparación de ORF filtrado fragmentos de la DNA
- Configurar digestión de la enzima de la restricción de 5 μg de vector purificado desde el vector de biblioteca pFILTER ORF añadiendo 10 U de BssHII y de incubación según protocolo del fabricante. Inactivar la enzima y la recopilación con 10 U de NheI.
- Carga de la DNA digerida en gel de agarosa al 1.5%, junto con una escalera de DNA 100 bp. Realizar una electroforesis corto en 5 V/cm durante 15 minutos o lo suficiente distinguir el borrón de transferencia de los fragmentos suprimidos y cortar la porción del gel que las contengan.
- Purificar el ADN inserto con un kit de extracción de gel de base de la columna y medir la concentración utilizando un espectrofotómetro UV.
-
Preparación de phagemid DNA
- Configurar la digestión de la enzima de la restricción de 5 μg de pDAN5 purificada27 en cuanto a los rellenos.
- Purificar el plásmido digerido por la corriente ADN digerido en un gel de agarosa de 0,75% y extracto de gel con un juego basado en la columna.
- Medir la concentración utilizando un espectrofotómetro UV. Las muestras pueden almacenarse a-20 ° C hasta su uso.
-
Recolección, transformación y ligadura de la biblioteca
- Realizar la ligadura y la transformación como se describe para pFILTER vector.
- Transformado de placa DH5αF' 150 mm 2xYT placas de agar suplementado con ampicilina μg/mL 100 e incubar durante una noche a 30 ° C.
- Diluciones de la biblioteca en placas de agar de 2xYT 100 mm suplementado con ampicilina 100 de μg/mL para determinar el tamaño de la biblioteca de la placa.
- Realizar la validación de la biblioteca por PCR de clones al azar escogidos como se describe en el paso 1.4.
- Recoge la biblioteca phagemid-ORF de la cosecha de bacterias de las placas de 150 mm, mezclar bien, complementarlas con 20% de glicerol estéril y conservar a-80 ° C en pequeñas alícuotas.
- Purificar el DNA plasmídico de una alícuota de la biblioteca utilizando un kit de extracción de plásmidos basado en la columna, siguiendo las instrucciones del fabricante.
- Medir la concentración en el espectrofotómetro de UV. Las muestras pueden conservarse a-20 ° C hasta ser utilizadas para la caracterización de NGS.
3. fago biblioteca preparación y procedimiento de selección
- Producción de fago
- Diluir una alícuota de existencias de la biblioteca de phagemid en 10 mL de 2xYT líquido caldo suplementado con ampicilina μg/mL 100 para tener un OD600 nm = 0.05.
- Crece la biblioteca diluida en un matraz estéril 5 - 10 veces mayor que el volumen original, a 37 ° C con agitación a 220 rpm hasta que alcances OD600 nm = 0.5.
- Infectar a las bacterias con fago del ayudante (por ejemplo M13K07) en una multiplicidad de infección de 20:1. Dejar a 37 ° C por 45 min con agitación ocasional (cada 10 minutos).
- Centrifugue las bacterias a 4000 x g por 10 min a temperatura ambiente. Deseche el sobrenadante y resuspender el pellet de bacterias en 40 mL de 2xYT líquido caldo suplementado con 100 μg/mL ampicilina y kanamicina μg/mL 50 crecimiento a 28 ° C con agitación a 220 rpm para pasar la noche.
- Al día siguiente, las bacterias de centrifugar a 4000 x g por 20 min a 4 ° C. Recoger el sobrenadante que contiene los phages.
- PEG-precipitación de fagos.
- Añadir 1/5 volumen de 0,22 μm filtrado PEG/NaCl solución (20% w/v PEG 6000, 2.5 M NaCl) a los phages despejados e incubar en hielo durante 30-60 minutos.
Nota: Solución se convirtió en humo después de pocos minutos, indicando una precipitación exitosa phage. La turbidez de la solución aumentará el tiempo de incubación. - Centrifugar a 4000 x g durante 15 min a 4 ° C. Se formará una pelotilla pequeña blanca de fagos.
- Resuspender en 1 mL de PBS estéril. Transferir a tubo de 1.5 mL y centrifugar a 4 ° C por 10 min a máxima velocidad para eliminar las bacterias contaminantes. Se forma un sedimento marrón.
- Transferir fagos que contienen sobrenadante a un tubo nuevo. Mantenga phages en hielo para sucesiva selección titulación y fago.
- Añadir 1/5 volumen de 0,22 μm filtrado PEG/NaCl solución (20% w/v PEG 6000, 2.5 M NaCl) a los phages despejados e incubar en hielo durante 30-60 minutos.
- Valoración de fago
- Preparar diluciones seriadas de la solución de fago. Coloque 10 μl de la solución de fago en 990 μl de PBS para obtener la dilución-2 10. Diluir otra vez esta preparación y de esta obtener una dilución 10-6 10-4 .
- Crecer DH5αF' células de las bacterias en medio líquido de 2xYT a 37 ° C con agitación hasta OD600 nm = 0.5 se alcanza. Transfiera 1 mL de la bacteria preparada en tubo de 1,5 mL e inmediatamente infectar con 1 μl de la dilución 10-4 del phage. Incubar sin agitación a 37 ° C por 45 min repetir el mismo procedimiento para la dilución de 10-6 .
- Diluciones de la placa de la bacteria infectada en la placa de 100 mm 2xYT. Poner la placa a 30 ° C durante la noche.
- Placa de 100 μl de infectado DH5αF' en una placa de agar de 2xYT suplementado con ampicilina 100 de μg/mL para comprobar la ausencia de contaminación en la preparación.
- El día después de contar el número de colonias y calcular el título del fago. Título expresa como número de phages/mL. Título esperado es de 1012 y 13 de phages/mL.
- Selección de fagos
-
Selección de fagos utilizando como cebo purificado anticuerpos
- Saturar phages por dilución 200 μL de la preparación de fagos en un volumen igual de PBS - 4% descremada e incubar durante 1 h a temperatura ambiente en rotación lenta. Este paso permite bloqueo de fagos para el atascamiento no específico. Transferencia 30 μl de proteína G granos magnéticos recubiertos a un tubo de 1,5 mL.
- Lavar dos veces como sigue: agregar 500 μl de PBS, incubar en una rueda en rotación lenta por 2 min a temperatura ambiente, sacar los granos a un lado del tubo usando un imán y eliminar el sobrenadante.
- Incubar los phages saturados con los granos lavados durante 30 min a temperatura ambiente con rotación lenta.
- Sacar los granos a un lado usando un campo magnético. Recoger el sobrenadante que contiene los phages para la etapa de selección.
- Preparar granos magnéticos al realizar el paso anterior, por conjugar los anticuerpos purificados. Lavar 30 μl de proteína G granos magnéticos recubiertos como se describió anteriormente. Diluir 10 μg de anticuerpos purificados en 500 μl de PBS, añadir a los granos lavados e incube en rotación lenta a temperatura ambiente durante 45 min lavado dos veces con PBS.
Nota: Realizar dos preparaciones diferentes de granos magnéticos: uno con anticuerpos de interés y con control, por ejemplo, anticuerpos purificaron de donantes sanos. La secuencia de antígenos con anticuerpos se restan en la etapa de análisis de las salidas de control. Por otra parte, bolas magnéticas con anticuerpos de control pueden utilizarse para realizar un paso previo claro de los phages (seguir el protocolo para la incubación con conjugado sin granos). - Selección de fagos: llamar a cuentas a un lado del tubo utilizando un imán, quitar el último lavado, agregar phages e incube con rotación lenta a temperatura ambiente durante 90 min lavar 5 veces con 500 μl de Tween-20 de PBS-0.1% y 5 veces con PBS.
- Eluir phages consolidados, que representa la salida de la selección, mezclando los granos con 1 mL de DH5αF' cultivados en OD600 = 0,5. Incubar las bacterias con los granos durante 45 min a 37 ° C con agitación ocasional (cada 10 minutos). Placa de la salida de una placa de agar de 2xYT 150 mm suplementada con ampicilina 100 de μg/mL.
- Placa de 100 μl de no diluido y de diferentes diluciones de la salida (10-1 a 10-5) para realizar la valoración. El día después de recoger las bacterias de las placas de 150 mm añadiendo 3 mL de medio 2xYT frescos y cosecha con un raspador estéril, mezclar bien, complementar con el 20% de glicerol estéril y almacenar a-80 ° C en alícuotas pequeñas.
- Crecer una alícuota para realizar a una segunda ronda de selección. Repita todo el procedimiento de barrido como se describió anteriormente, excepto en las condiciones de lavado. En este caso lavar 10 veces con PBS - 1% Tween-20 (Vierta la solución en el tubo y vaciar inmediatamente). Luego agregar 500 μl de PBS e incubar en rotación a temperatura ambiente por 10 minutos realizar otros 10 lavados con PBS. Proceder con el paso de elución en cuanto a la primera ronda de selección.
- Extraer ADN de plásmido de una alícuota de la salida usando un juego basado en la columna, siguiendo las instrucciones del fabricante. Plásmido de tienda a-20 ° C hasta que se utilizará para secuenciación profunda.
-
Selección de fagos utilizando como cebo de proteínas recombinantes
- Saturar phages por dilución 200 μL de la preparación de fagos en un volumen igual de PBS - 4% descremada e incubar durante 1 h a temperatura ambiente en rotación lenta.
- Añada 100 μl de granos magnéticos de estreptavidina. Incubar durante 1 h a temperatura de ambiente para seleccionar enlace de estreptavidina phages. Retire los phages de la estreptavidina-limite mediante la elaboración de las cuentas a un lado utilizando un imán. Tomar el sobrenadante del paso anterior y agregar proteína biotinilada (en una concentración de 100-550 nM) e incubar en un rotor a temperatura ambiente por 30 min a 1 h.
- Preparar granos magnéticos: al realizar el paso anterior, lavar 100 μl de granos estreptavidina-magnéticos con PBS, resuspender en PBS 2% descremada e incubar con rotación a temperatura ambiente durante 30 min a 1 h.
- Selección de fagos: sacar los granos a un lado del tubo utilizando un imán, quitar PBS - 2% leche y suspender cuentas con mezcla de proteínas del fago. Incubar con rotación lenta a temperatura ambiente durante 90 minutos.
- Sacar los granos a un lado del tubo utilizando un imán, deseche el sobrenadante y lavar cuidadosamente cinco veces con 500 μl de PBS 0,1% Tween-20. Realizar la elución como se describe en la sesión anterior.
-
Selección de fagos utilizando como cebo purificado anticuerpos
4. el fago biblioteca secuenciación profunda plataforma (figura 3)
-
Insertos de DNA de pFILTER-ORF-biblioteca, pDAN5-ORF-biblioteca o bibliotecas de fagos seleccionados
- Descongelar una alícuota de la biblioteca, cuantificar mediante el uso de un espectrofotómetro, recupera las inserciones de ADN por amplificación con iniciadores específicos.
Nota: Los iniciadores utilizados para rescatar a los insertos están ligados en su extremo 5' secuencias de adaptadores, lo que permite la indexación de direcciones sucesivas de las piscinas de amplicones obtenidos y la secuencia directa de los insertos de ADN recuperados mediante el uso de los secuenciadores. Su secuencia es en la Tabla de materiales. Los adaptadores están indicados en negrita, y los cebadores específicos se indican en cursiva. - Utilizar 2,5 μl de la biblioteca (pFILTER/phagemid/seleccionado-phage) como plantilla de la DNA para una reacción de PCR.
- Utilizar el siguiente programa: 95 ° C por 3 min; 25 ciclos de 95 ° C por 30 s, 55 ° C por 30 s, 72 ° C por 30 s; 72 ° C para el asimiento de 5 min a 4 ° C.
Nota: en este punto se recomienda ejecutar 1 μl del producto PCR en un Bioanalizador o TapeStation para verificar el tamaño de los amplicones y verificar que estén en el rango correcto.
- Descongelar una alícuota de la biblioteca, cuantificar mediante el uso de un espectrofotómetro, recupera las inserciones de ADN por amplificación con iniciadores específicos.
-
Limpiar el PCR
- Poner las bolas magnéticas (por ejemplo, Aonang) a temperatura ambiente. Transferir todo el producto PCR desde el tubo PCR en un tubo de 1,5 mL. Vórtice las bolas magnéticas para 30 s para asegurarse de que los granos están uniformemente dispersos. Añadir 20 μl de granos magnéticos a cada tubo que contiene el producto PCR, mezclar mediante pipeteo suave. Incubar a temperatura ambiente sin agitar durante 5 minutos.
- Coloque la placa en un soporte magnético por 2 min o hasta que haya desaparecido el sobrenadante. Con los productos de la polimerización en cadena en el soporte magnético, retire y descarte el sobrenadante.
- Lavar los granos con etanol de 80% recién preparada, con los productos PCR en el soporte magnético, como sigue: agregar 200 μL de etanol al 80% recién preparada para cada muestra bien; Incube la placa en el soporte magnético para 3 s; cuidadosamente retire y descarte el sobrenadante.
- Realizar un segundo lavado con etanol, con los productos PCR en el soporte magnético; al final del segundo lavado cuidadosamente retire el etanol y los granos de secar al aire durante 10 minutos.
- Eliminar los productos PCR del soporte magnético, agregar 17.5 μl de 10 mM de Tris de pH 8.5 a cada tubo, pipetee suavemente hacia arriba y hacia abajo 10 veces, asegúrese de que los granos son completamente suspendidos. Incubar a temperatura ambiente durante 2 minutos.
- Coloque el tubo en el soporte magnético para 2 min o hasta que haya desaparecido el sobrenadante, con cuidado de transferencia 15 μl del sobrenadante que contiene los productos PCR purificados a un nuevo tubo de 1,5 mL. Almacenar los productos PCR purificados de-15 ° C a-25 ° C durante hasta una semana si no procede inmediatamente a la PCR de índice.
-
Índice de polimerización en cadena
Nota: Después de limpiar el PCR, realizar PCR de índice. Utilice el kit de Nextera XT índice; así será posible la secuencia de las librerías indexadas doble resultantes dentro de multiplexado funciona Illumina.- Transferir todos los 15 μL que contiene cada producto purificado en un nuevo tubo PCR y configurar la siguiente reacción que contiene: 15 μl del producto de amplicón purificado, 5 μl de índice cartilla 1 y 5 μl de índice cartilla 2, 25 μl de la mezcla de x PCR 2; volumen final de 50 μl.
- Realizar PCR en un termociclador con el siguiente programa: 95 ° C por 3 min, 8 ciclos de 95 ° C por 30 s, 55 ° C por 30 s, 72 ° C por 30 s; 72° C por 5 min, después llevar a cabo a 4 ° C.
-
PCR de limpiar 2
- Seguir el mismo protocolo descrito en la sección 4.2 para PCR limpiar con los siguientes cambios: en el primer paso añadir 56 μl de granos magnéticos a cada 50 μl del producto de PCR.
- Suspender los granos en 27,5 μl de 10 mM Tris, pH 8.5 en el último paso de purificación y transferir 25 μl a un tubo nuevo (esta es la purificada biblioteca final lista para la cuantificación y luego secuenciación).
- Almacenar la placa a-15 ° C a-25 ° C durante hasta una semana si no proceder a la cuantificación de la biblioteca.
-
Evaluación cualitativa y cuantitativa de la biblioteca de la secuencia
- Después de la purificación, ejecute 1 μl de un 1:10 dilución de la biblioteca final en un equipo bioanalyzer para verificar el tamaño y cuantificar seleccionando la región de la traza definitiva de la biblioteca.
- Paralelamente realizar la cuantificación de la biblioteca por PCR en tiempo Real mediante un kit de cuantificación de la biblioteca según el protocolo del fabricante.
-
Bibliotecas de secuenciación
- Piscina las librerías indexadas duales producidas junto con otras bibliotecas de doble secuencia indexadas. Secuencia de que este tipo de biblioteca generando mucho Lee, por lo menos 250 extremo apareado bp usando el Hiseq2500 o los instrumentos de MiSeq para obtener en el primer caso 250bp PE Lee y en el segundo caso 300 bp PE Lee.
5. Análisis de datos bioinformática mediante la herramienta de Web interactoma-Seq
- Analizar la Lee originada de secuenciación biblioteca pFILTER/phagemid/seleccionado-fago con el gasoducto de análisis de datos interactoma-seq. La herramienta web está disponible gratuitamente en la siguiente dirección: http://interactomeseq.ba.itb.cnr.it/
Resultados
El enfoque de filtrado es esquematizado en la figura 1. Puede utilizarse cada tipo de ADN intronless. En la figura 1A se representa la primera parte de lo filtrado: después de la carga en un gel de agarosa o un equipo bioanalyzer, una buena fragmentación de la DNA de interés aparece como un borrón de transferencia de los fragmentos con una distribución de longitud en el tamaño deseado de 150-750 bp. Se da una imagen de representante virtual gel de la DNA fragmentada obtenida. Fragmentos en el gel de agarosa luego recuperados, reparado final y fosforiladas y entonces clonados en un vector previamente blunted pFILTER para crear una biblioteca de fragmentos de ADN al azar. Realizar cada paso del procedimiento de clonación en condiciones óptimas es necesaria para obtener buena calidad biblioteca con una cobertura total del ADN en estudio.
En la figura 1B se representa el enfoque de filtrado: la biblioteca se cultiva en presencia de cloranfenicol (pFILTER resistencia) solo o cloranfenicol y ampicilina para seleccionar las colonias que contiene el ORF. Sólo las colonias con un fragmento de ADN correspondiente a un ORF producen una β-lactamasa funcional y sobreviven cuando la selección antibiótica está presente. Figura 1 muestra cómo aumenta la presión selectiva permite selección de carpeta buena ORFs versus carpeta pobres unos. El resultado esperado es una disminución del tamaño de la biblioteca de unos 20-fold. Mayor número de clones sobrevivientes indica insuficiente presión selectiva.
Fragmentos ORF pueden ser fácilmente recuperados de la biblioteca filtrada para su posterior aplicación; estudios de interacción nuestra estrategia aprovecha la tecnología de exhibición phage. En la figura 2, se representan los principales pasos de construcción de biblioteca de fago: una biblioteca adecuada se prepara por reducción de filtrado fragmentos del vector pFILTER y volver a clonar en un plásmido phagemid en fusión con la secuencia de codificación para el fago capside proteína g3p. Una vez infectado con un fago del ayudante, la presencia del vector en las células de las bacterias permite la producción de partículas de fago mostrando productos g3p ORF en su superficie por lo que la biblioteca de filtrado disponibles para la selección de phage display y más Análisis.
Todas las bibliotecas son profundamente analizadas por NGS, así como las salidas de las selecciones de fago, como se muestra en la segunda parte de la figura 3. Fragmentos de ADN son rescatados del cultivo colonias por amplificación por PCR con oligonucleótidos específicos de recocido en la columna vertebral del plásmido y llevar adaptadores específicos para la secuenciación. NGS se realiza y Lee luego es analizado con la herramienta de web de análisis de datos interactoma-Seq.
En la figura 4 hemos informado una representación esquemática del procedimiento de selección de una biblioteca ORF filtrada phage display. La selección en este ejemplo se realiza mediante el uso de anticuerpos presentes en el suero de pacientes afectados por distintas patologías (es decir, patologías infecciosas, patologías autoinmunes, cáncer). En este caso la biblioteca phage directamente interactúa con los anticuerpos presentes en los sueros de los pacientes y de esta manera posibles antígenos específicos pueden enriquecerse porque son reconocidas por los anticuerpos específicos de enfermedad. En este tipo de experimento, la biblioteca es también selecciona generalmente usando sueros de control de pacientes sanos para tener una señal de fondo a utilizar para sucesivos procedimientos de comparación y la normalización.
Selecciones se realizan utilizando sueros del mismo tipo de pacientes que generalmente se agrupan en diferentes piscinas con el fin de reducir la variabilidad interindividual del título del anticuerpo del suero. Cada piscina se utiliza independientemente para dos o tres rondas consecutivas de la selección, para enriquecer la biblioteca de clones inmunológico reactivo específicos para la patología en estudio. Anticuerpos sets prueba se incuban con phages de la biblioteca, complejos inmunes son recuperados por magnetics-perlas de proteína A cubierto y fagos enlazados se eluyen por procedimientos estándar. Los ciclos de selección se realizan con el aumento del lavado y vinculante de rigor.
Las lecturas generadas por NGS pueden analizarse utilizando la herramienta de web interactoma Seq específicamente desarrollada para manejar este tipo de datos. Interactoma Seq workflow de análisis de datos se compone de cuatro pasos secuenciales que, a partir de lecturas de secuencia de la materia prima, genera la lista de dominios putativos con anotaciones genómicos (figura 5A). En el primer paso de entrada (figura 5A - caja roja), interactoma Seq verifica si son correctamente el formato de los archivos de entrada (lecturas materia prima, secuencia del genoma de referencia, lista de anotación). En el segundo paso de PREPROCESAMIENTO (caja de lafigura 5A - naranja), datos de baja calidad la secuencia primero se recortan utilizando Cutadapt28 dependiendo de los resultados de calidad y se descarta los Lee con menos de 100 bases de longitud. En un paso posterior de alineación leer (caja de lafigura 5A - verde), las lecturas restantes se alinean con blastn29 a la secuencia del genoma que permite hasta un 5% de uniones mal hechas. Un archivo SAM se genera y sólo lee con puntuación de calidad superior a 30 (Q > 30) se procesan usando SAMtools30 y se convierte en un archivo BAM. Después de la alineación, Seq interactoma realiza la detección de dominios (figura 5A - caja azul), invocando Bedtools31 a filtro Lee superposición de al menos 80% de su longitud dentro de transcripciones; la cobertura, profundidad máxima y valores enfoque entonces se calculan por cada porción ORF cubierta mediante la asignación de lecturas. La cobertura representa el número total de lecturas asignadas a un gen; la profundidad es el número máximo de lecturas que abarcan una porción génica específica; el foco es un índice Obtenido de la relación entre la cobertura y la profundidad máxima, y puede variar entre 0 y 1. Cuando el foco es mayor que 0.8 y la cobertura es mayor que la cobertura promedio para todas las regiones de mapeo en el archivo BAM, la porción de CD se clasifica como un dominio putativo/epítopo. El último paso de la tubería del interactoma Seq es la salida (figura 5A - caja violeta), se genera una lista de dominios putativos en formato tabular separado. La tubería del interactoma Seq ha sido incluida en una herramienta web para permitir a usuarios sin conocimientos de programación ni bioinformática para realizar análisis del interactoma Seq a través de la interfaz gráfica y obtener sus resultados en un formato fácil y fácil de usar. Como se muestra en la figura 5B, se muestran los resultados de salida de un análisis usando JBrowse32 para permitir la visualización y exploración. Interactoma Seq genera pistas en el navegador de genoma correspondiente a dominios putativos detectados y proporciona también los clásicos diagramas de Venn para Mostrar intersecciones entre dominios putativos común enriquecidos por ejemplo en los experimentos de diferentes selecciones.

Figura 1: Descripción esquemática de los pasos principales para la construcción de la biblioteca de filtrado de ORF
A) ADN de origen diferente es sonicado y fragmentado en fragmentos al azar de la longitud de 150-750 bp. Fragmentos recuperados de gel y clonados como embotado en el vector pFILTER; B) paso filtrado usando β-lactamasa como reportero plegable. Vector que contiene fragmentos ORF no se selecciona negativamente en ampicilina mientras ORF fragmentos clonados permiten colonias creciendo; C) la aplicación de un incremento de la presión selectiva (concentración de ampicilina en medios de cultivo sólido de 0 a > 100 μg/mL) permiten la selección de los fragmentos mejores doblados. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Esquema Resumen de los pasos principales para la construcción de la biblioteca de fago
A) fragmentos de filtradas por ORF se recortan desde el vector filtrado utilizando enzimas de restricción específicas. Después de la recuperación y purificación, fragmentos son clonados en el vector phagemid y transformados; B) Biblioteca bacteriana phagemid está infectado con el fago del ayudante y, después de crecimiento durante la noche, phages son precipitados de PEG y recogidos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Bibliotecas ORF secuenciación
La secuencia se realiza en ambos la original ORF biblioteca seleccionada así como en la biblioteca de phage display; 1) en ambos casos se recuperan colonias cultivadas y ADN extraído; 2) fragmentos de ADN son recuperados por amplificación con iniciadores específicos ligados a adaptadores para secuenciación; 3-4) fragmentos son recuperados y profundos ordenado usando NGS; 5) los datos se analizan mediante el uso de la tubería del interactoma-Seq. Haga clic aquí para ver una versión más grande de esta figura.
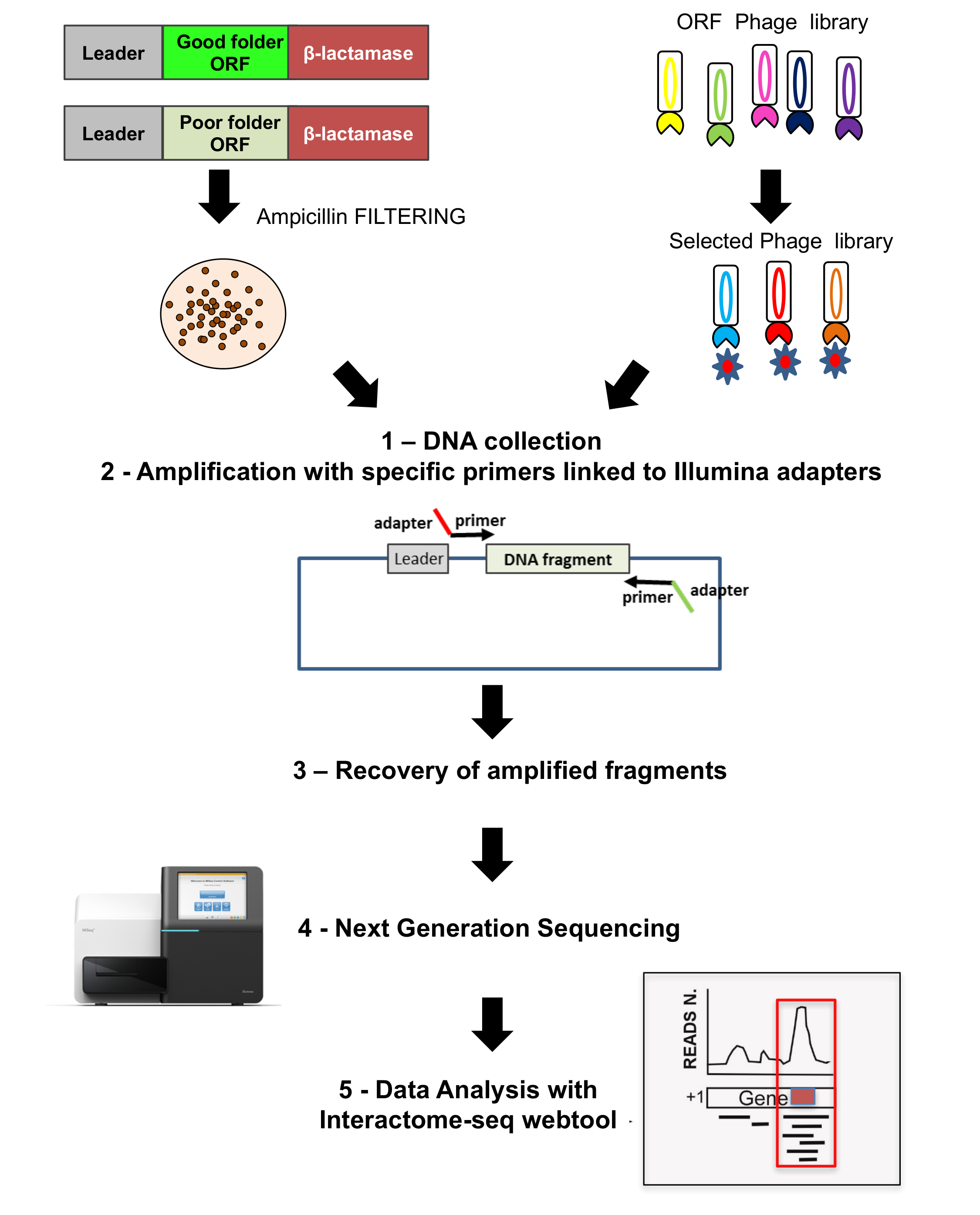{kind=link}

Figura 4: Esquema Resumen de la selección de la biblioteca utilizando anticuerpos de pacientes
Biblioteca de fago se utiliza para la selección contra anticuerpos de sueros de pacientes. Los anticuerpos se inmovilizan en granos magnéticos, el fago biblioteca captura y selección se lleva a cabo, se realizan tres ciclos de lavados y luego phages seleccionados son recuperados y utilizados para volver a infectar e. coli. Volver a infectados por e. coli las células se platean en presión selectiva (ampicilina 100 μg/mL). Se recuperan fragmentos ORF por amplificación y productos piscinas son luego secuenciados por NGS. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Esquema Resumen de análisis de biblioteca
A) representación del flujo de trabajo de análisis de datos, a partir de archivos raw FASTQ a las listas de dominios comentada final; B) esquemáticamente las entradas y salidas de la herramienta de web interactoma-Seq. Haga clic aquí para ver una versión más grande de esta figura.
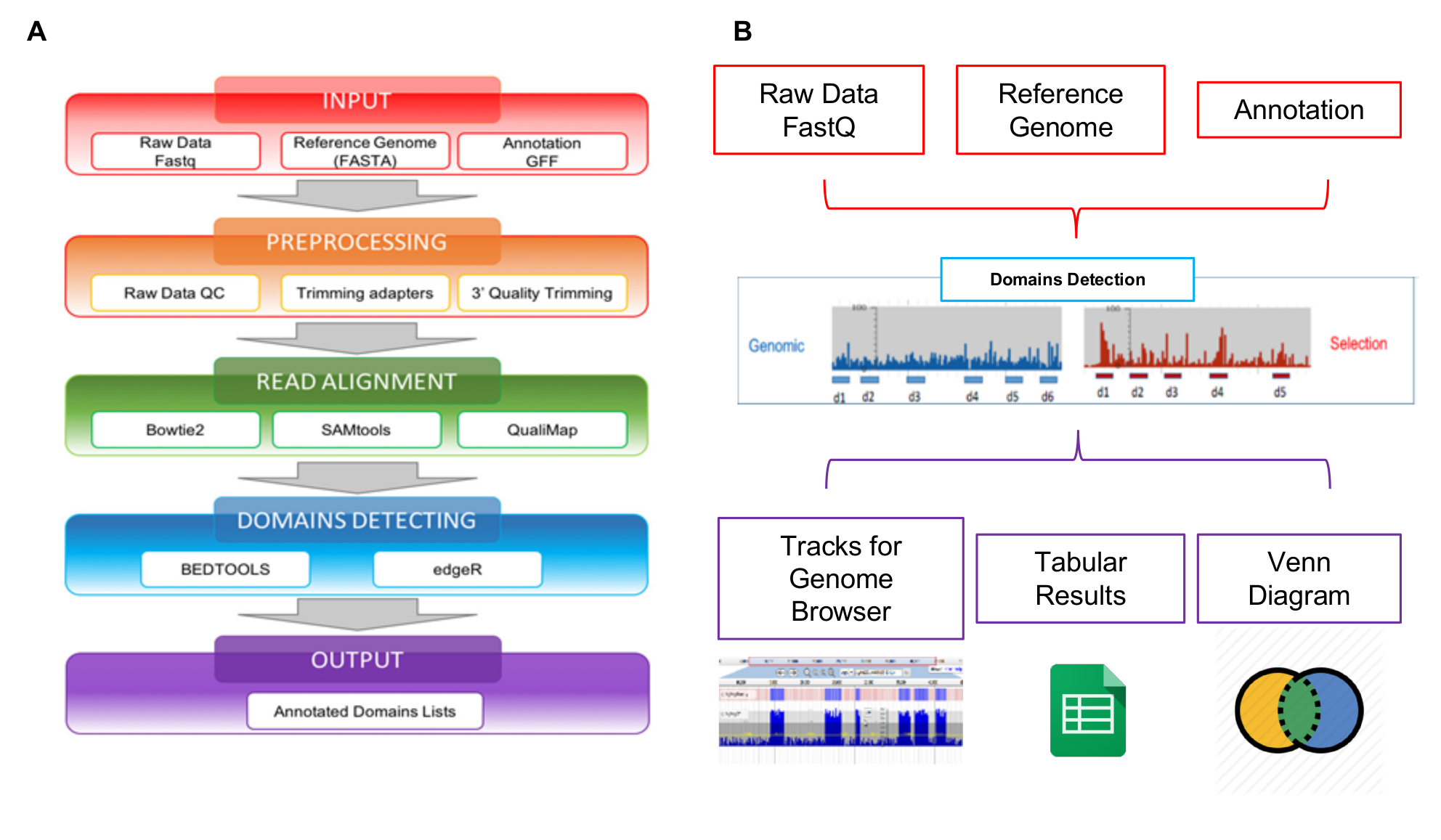{kind=link}
Discusión
La creación de una biblioteca alta calidad muy diversa ORFs filtrada es el primer paso crítico en todo el procedimiento ya que afectará a todos los pasos subsecuentes de la tubería.
Una característica importante de la ventaja de nuestro método es que cualquier fuente de ADN (intronless) (ADNc, ADN genómico, derivado de la PCR o ADN sintético) es conveniente para la construcción de la biblioteca. El primer parámetro que debe tenerse en cuenta es que la longitud de los fragmentos de ADN clonados en el vector pFILTER debe ofrecer una representación de toda la colección de los dominios de un genoma o un transcriptoma, el llamado "domainome". Hemos demostrado que dominios de la proteína pueden ser clonados con éxito, seleccionan y finalmente identificaron a partir de fragmentos de ADN con una longitud distribución que abarca desde 150 hasta 750 bp33,34, y esto está en consonancia con lo que se divulga en la literatura que muestra que más dominios de la proteína son de longitud aa 100 (con un rango de 50 a 200 aa)15.
ADN a partir de material se debe fragmentado en el rango de tamaño de la opción y posteriormente clonados en el vector de12 (pFILTER) filtrado. Durante estos pasos, se podrían evitar posibles sesgos maximizando la eficiencia de todos los pasos de la clonación las reacciones incluidas en el protocolo, en particular fragmento final-reparación y fosforilación. La preparación del vector es difícil y debe hacerse en condiciones óptimas, para evitar degradación de plásmido o contaminación por vectores sin digerir.
Una vez que se ha creado la biblioteca, se debe ser "filtrado" para conservar solamente los fragmentos doblados ORFs. Un parámetro clave para modular este paso es el selectivo presión aplicada que puede ser modificada según la severidad de la filtración deseada. Selección se realiza con ampicilina: cuanto mayor sea la concentración utilizada, menor será el número de colonias de bacterias transformadas capaces de sobrevivir. Esto refleja la capacidad del método de filtrado para seleccionar de bueno-versus pobres-carpeta ORFs34. Esta reducción en el número de clones es equilibrada por el aumento en la propiedades de los fragmentos seleccionados que dobla. Generalmente, la concentración de ampicilina debe ser suficiente para reducir a 1/20 el número de colonias de bacterias con respecto a las que podrían obtenerse creciendo la biblioteca en el cloranfenicol solo.
Validación de la biblioteca se realiza generalmente por amplificación por PCR de las colonias al azar escogidas y su secuencia. Amplificación por PCR de algunas colonias se sugiere para tener una estimación rápida de la calidad de la biblioteca: la longitud de las inserciones debe ser en el rango de 150-750 bp y diversas colonias deben presente insertos con diferentes tamaño que indica buena preparación de biblioteca en términos de variabilidad. Esta estrategia convencional de proyección, cuando se aplica como el único método para la validación de la biblioteca, no es completa y es mucho tiempo, permitiendo el análisis de un número limitado de colonias y tener una alta probabilidad de perder la mayoría de los clones importantes. Nuestro enfoque se basa en la secuencia profunda de la biblioteca, esto proporciona información completa sobre la biblioteca diversidad y abundancia y asignación precisa de cada uno de los fragmentos seleccionados.
La implementación de tecnología NGS con el enfoque de filtrado aumenta la profundidad del análisis en varios órdenes de magnitud. Recientemente, hemos optimizado el protocolo para las bibliotecas ORF de la secuencia mediante el uso de la plataforma Illumina y desarrollado una herramienta de web específica para el análisis de datos que hacen que el análisis de este tipo de datos para cada usuario sin cualquier bioinformática conocimientos de programación.
La biblioteca "per se" es un "instrumento universal" y puede ser explotada en diferentes contextos para la expresión de la proteína o selección. Nuestro enfoque metodológico se basa en la transferencia de la ORFeome producido en un contexto de exhibición phage. Fragmentos de proteína se expresan en la superficie de fagos y llegó a ser convenientes para la posterior selección.
Esto se hace por rescatar el ORFs filtradas de la biblioteca pFILTER por digestión con enzimas de restricción específicas y repetir los clonación en un vector compatible phagemid permitiendo su fusión con el g3p de proteínas del fago.
Una vez creada la biblioteca phagemid-ORF, puede ser utilizado para la selección contra diferentes objetivos, como una supuesta Unión proteína10 anticuerpos purificados35,36 como se describe aquí. Desde las partículas phage display en su superficie los ORFs filtradas, esto da lugar a un procedimiento de selección más eficaz debido a la ausencia de no mostrar clones que generalmente superará lo.
Después de la selección de la biblioteca ORF de phage display, los clones de salida pueden ser secuenciados y analizados con la misma tubería. NGS puede proporcionar una completa y ranking estadísticamente significativa de los más frecuentemente seleccionados ORFs y esto permite la identificación de las proteínas sobre todo interactuando con el cebo utilizado. Ante la presencia de muchas versiones diferentes de cada dominio difieren por pocos aminoácidos, la superposición entre diferentes clones secuenciados también identifica el mínimo fragmento/dominio que muestra propiedades de enlace. Por último, gracias al acoplamiento de genotipo y fenotipo de información en la biblioteca de fago, una vez identificados los dominios de la opción, la secuencia del ADN puede fácilmente rescatar de la biblioteca para más estudios, in vitro e in vivo validación y caracterización.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Este trabajo fue financiado por una subvención del Ministerio de Educación Italiano y la Universidad (2010P3S8BR_002 CP).
Materiales
| Name | Company | Catalog Number | Comments |
| Sonopuls ultrasonic homogenizer | Bandelin | HD2070 | or equivalent |
| GeneRuler 100 bp Plus DNA Ladder | Thermo Scientific | SM0321 | or equivalent |
| GeneRuler 1 kb DNA Ladder | Thermo Fisher Scientific | SM0311 | or equivalent |
| Molecular Biology Agarose | BioRad | 161-3102 | or equivalent |
| Green Gel Plus | Fisher Molecular Biology | FS-GEL01 | or equivalent |
| 6x DNA Loading Dye | Thermo Fisher Scientific | R0611 | or equivalent |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | or equivalent |
| Quick Blunting Kit | New England Biolabs | E1201S | |
| NanoDrop 2000 UV-Vis Spectrophotometer | Thermo Fisher Scientific | ND-2000 | |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | 4368813 | |
| Streptavidin Magnetic Beads | New England Biolabs | S1420S | or equivalent |
| QIAquick PCR purification Kit | Qiagen | 28104 | or equivalent |
| EcoRV | New England Biolabs | R0195L | |
| Antarctic Phosphatase | New England Biolabs | M0289S | |
| T4 DNA Ligase | New England Biolabs | M0202T | |
| Sodium Acetate 3M pH5.2 | general lab supplier | ||
| Ethanol for molecular biology | Sigma-Aldrich | E7023 | or equivalent |
| DH5aF' bacteria cells | Thermo Fisher Scientific | ||
| 0,2 ml tubes | general lab supplier | ||
| 1,5 ml tubes | general lab supplier | ||
| 0,1 cm electroporation cuvettes | Biosigma | 4905020 | |
| Electroporator 2510 | Eppendorf | ||
| 2x YT medium | Sigma-Aldrich | Y1003 | |
| Ampicillin sodium salt | Sigma-Aldrich | A9518 | |
| Chloramphenicol | Sigma-Aldrich | C0378 | |
| DreamTaq DNA Polymerase | Thermo Fisher Scientific | EP0702 | |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | N0447S | |
| 96-well thermal cycler (with heated lid) | general lab supplier | ||
| 150 mm plates | general lab supplier | ||
| 100 mm plates | general lab supplier | ||
| Glycerol | Sigma-Aldrich | G5516 | |
| BssHII | New England Biolabs | R0199L | |
| NheI | New England Biolabs | R0131L | |
| QIAprep Spin Miniprep Kit | Qiagen | 27104 | or equivalent |
| M13KO7 Helper Phage | GE Healthcare Life Sciences | 27-1524-01 | |
| Kanamycin sulfate from Streptomyces kanamyceticus | Sigma-Aldrich | K1377 | |
| Polyethylene glycol (PEG) | Sigma-Aldrich | P5413 | |
| Sodium Cloride (NaCl) | Sigma-Aldrich | S3014 | |
| PBS | general lab supplier | ||
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher Scientific | 10003D | or equivalent |
| MagnaRack Magnetic Separation Rack | Thermo Fisher Scientific | CS15000 | or equivalent |
| Tween 20 | Sigma-Aldrich | P1379 | |
| Nonfat dried milk powder | EuroClone | EMR180500 | |
| KAPA HiFi HotStart ReadyMix | Kapa Biosystems, Fisher Scientific | 7958935001 | |
| AMPure XP beads | Agencourt, Beckman Coulter | A63881 | |
| Nextera XT dual Index Primers | Illumina | FC-131-2001 or FC-131-2002 or FC-131-2003 or FC-131-2004 | |
| MiSeq or Hiseq2500 | Illumina | ||
| Spectrophotomer | Nanodrop | ||
| Agilent Bioanalyzer or TapeStation | Agilent | ||
| Forward PCR primer | general lab supplier | 5’ TACCTATTGCCTACGGCA GCCGCTGGATTGTTATTACTC 3’ | |
| Reverse PCR primer | general lab supplier | 5’ TGGTGATGGTGAGTACTA TCCAGGCCCAGCAGTGGGTTTG 3’ | |
| Forward primer for NGS | general lab supplier | 5’ TCGTCGGCAGCGTCAGA TGTGTATAAGAGACAGGCA GCAAGCGGCGCGCATGC 3’; | |
| Reverse primer for NGS | general lab supplier | 5’ GTCTCGTGGGCTCGGAGA TGTGTATAAGAGACAGGGG ATTGGTTTGCCGCTAGC 3’; |
Referencias
- Loman, N. J., Pallen, M. J. Twenty years of bacterial genome sequencing. Nat Rev Microbiol. 13 (12), 787-794 (2015).
- Jones, C. E., Brown, A. L., Baumann, U. Estimating the annotation error rate of curated GO database sequence annotations. BMC Bioinformatics. 8 (1), 170 (2007).
- Andorf, C., Dobbs, D., Honavar, V. Exploring inconsistencies in genome-wide protein function annotations: a machine learning approach. BMC Bioinformatics. 8 (1), 284 (2007).
- Wong, W. -. C., Maurer-Stroh, S., Eisenhaber, F. More Than 1,001 Problems with Protein Domain Databases: Transmembrane Regions, Signal Peptides and the Issue of Sequence Homology. PLoS Comput Biol. 6 (7), e1000867 (2010).
- Bioinformatics, B., et al. Identification and correction of abnormal, incomplete and mispredicted proteins in public databases. BMC Bioinformatics. 9 (9), (2008).
- Phizicky, E., Bastiaens, P. I. H., Zhu, H., Snyder, M., Fields, S. Protein analysis on a proteomic scale. Nature. 422 (6928), 208-215 (2003).
- DiDonato, M., Deacon, A. M., Klock, H. E., McMullan, D., Lesley, S. A. A scaleable and integrated crystallization pipeline applied to mining the Thermotoga maritima proteome. J Struct Funct Genomics. 5 (1-2), 133-146 (2004).
- Nordlund, P., et al. Protein production and purification. Nat Methods. 5 (2), 135-146 (2008).
- Zacchi, P., Sblattero, D., Florian, F., Marzari, R., Bradbury, A. R. M. Selecting open reading frames from DNA. Genome Res. 13 (5), 980-990 (2003).
- Di Niro, R., et al. Rapid interactome profiling by massive sequencing. Nucleic Acids Res. 38 (9), e110 (2010).
- Gourlay, L. J., et al. Selecting soluble/foldable protein domains through single-gene or genomic ORF filtering: Structure of the head domain of Burkholderia pseudomallei antigen BPSL2063. Acta Crystallogr Sect D Biol Crystallogr. 71 (Pt 11), 2227-2235 (2015).
- D'Angelo, S., et al. Filtering "genic" open reading frames from genomic DNA samples for advanced annotation. BMC Genomics. 12 (Suppl 1), S5 (2011).
- D'Angelo, S., et al. Profiling celiac disease antibody repertoire. Clin Immunol. 148 (1), 99-109 (2013).
- Robinson, M. D., McCarthy, D. J., Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26 (1), 139-140 (2009).
- Heger, A., Holm, L. Exhaustive enumeration of protein domain families. J Mol Biol. 328 (3), 749-767 (2003).
- Zacchi, P., Sblattero, D., Florian, F., Marzari, R., Bradbury, A. R. M. Selecting open reading frames from DNA. Genome Res. 13 (5), 980-990 (2003).
- Faix, P. H., Burg, M. A., Gonzales, M., Ravey, E. P., Baird, A., Larocca, D. Phage display of cDNA libraries: Enrichment of cDNA expression using open reading frame selection. Biotechniques. 36 (6), 1018-1029 (2004).
- Patrucco, L., et al. Identification of novel proteins binding the AU-rich element of α-prothymosin mRNA through the selection of open reading frames (RIDome). RNA Biol. 12 (12), 1289-1300 (2015).
- Collins, M. O., Choudhary, J. S. Mapping multiprotein complexes by affinity purification and mass spectrometry. Curr Opin Biotechnol. 19 (4), 324-330 (2008).
- Suter, B., Kittanakom, S., Stagljar, I. Two-hybrid technologies in proteomics research. Curr Opin Biotechnol. 19 (4), 316-323 (2008).
- Nakai, Y., Nomura, Y., Sato, T., Shiratsuchi, A., Nakanishi, Y. Isolation of a Drosophila gene coding for a protein containing a novel phosphatidylserine-binding motif. J Biochem. 137 (5), 593-599 (2005).
- Deng, S. J., et al. Selection of antibody single-chain variable fragments with improved carbohydrate binding by phage display. J Biol Chem. 269 (13), 9533-9538 (1994).
- Danner, S., Belasco, J. G. T7 phage display: A novel genetic selection system for cloning RNA-binding proteins from cDNA libraries. Proc Natl Acad Sci. 98 (23), 12954-12959 (2001).
- Gargir, A., Ofek, I., Meron-Sudai, S., Tanamy, M. G., Kabouridis, P. S., Nissim, A. Single chain antibodies specific for fatty acids derived from a semi-synthetic phage display library. Biochim Biophys Acta - Gen Subj. 1569 (1-3), 167-173 (2002).
- Patrucco, L., et al. Identification of novel proteins binding the AU-rich element of α-prothymosin mRNA through the selection of open reading frames (RIDome). RNA Biol. 12 (12), 1289-1300 (2015).
- Ausubel, F. M., et al. Current Protocols in Molecular Biology. Mol Biol. 1 (2), 146 (2003).
- Sblattero, D., Bradbury, A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat Biotechnol. 18, 75-80 (2000).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 17 (1), 10 (2011).
- Camacho, C., et al. BLAST+: architecture and applications. BMC Bioinformatics. 10 (1), 421 (2009).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Quinlan, A. R. BEDTools: The Swiss-Army tool for genome feature analysis. Curr Protoc Bioinforma. , (2014).
- Skinner, M. E., Uzilov, A. V., Stein, L. D., Mungall, C. J., Holmes, I. H. JBrowse: A next-generation genome browser. Genome Res. 19 (9), 1630-1638 (2009).
- Gourlay, L. J., et al. Selecting soluble/foldable protein domains through single-gene or genomic ORF filtering: Structure of the head domain of Burkholderia pseudomallei antigen BPSL2063. Acta Crystallogr Sect D Biol Crystallogr. 71, 2227-2235 (2015).
- D'Angelo, S., et al. Filtering "genic" open reading frames from genomic DNA samples for advanced annotation. BMC Genomics. 12 (Suppl 1), S5 (2011).
- Di Niro, R., et al. Characterizing monoclonal antibody epitopes by filtered gene fragment phage display. Biochem J. 388 (Pt 3), 889-894 (2005).
- D'Angelo, S., et al. Profiling celiac disease antibody repertoire. Clin Immunol. 148 (1), 99-109 (2013).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados