
Method Article
Interaktom-Seq: Ein Protokoll zur Domainome Bibliotheksbau, Validierung und Auswahl von Phagen-Display und Next Generation Sequencing
In diesem Artikel
Zusammenfassung
Die beschriebenen Protokolle ermöglichen Aufbau, Charakterisierung und Auswahl (gegen das Ziel der Wahl) einer "Domainome"-Bibliothek, die aus jeder DNA-Quelle hergestellt. Dies wird erreicht durch eine Forschungs-Pipeline, die verschiedenen Technologien kombiniert: Phagen-Display, eine klappbare Reporter und Sequenzierung der nächsten Generation mit einem Web-Tool für die Datenanalyse.
Zusammenfassung
Klappbare Reporter sind Proteine mit leicht erkennbaren Phänotypen, wie z. B. Antibiotika-Resistenz, dessen Falten und Funktion beeinträchtigt wird, wenn schlecht Faltung von Proteinen oder zufällige open Reading Frames verschmolzen. Wir haben eine Strategie entwickelt, kann mithilfe von TEM-1 β-Lactamase (das Enzym überträgt die Ampicillin-Resistenz) auf genomischer Ebene, Sammlungen von korrekt gefaltete Protein Domains wählen wir aus der Codierung Teil der DNA von jedem intronless Genom. Die PROTEINFRAGMENTE erhalten durch diesen Ansatz, der so genannte "Domainome", werden gut ausgedrückt und löslich, so dass sie für strukturelle und funktionelle Studien sein.
Von Klonen und die Anzeige "Domainome" direkt in einem Phagen-Display-System, haben wir zeigten, dass spezifisches Protein Domains mit der gewünschten Bindeeigenschaften (z. B. an andere Proteine oder Antikörper), wählen dadurch wesentlich kann experimentellen Daten für gen-Annotation oder Antigen-Identifikation.
Die Identifikation der meisten angereicherten Klone in einer ausgewählten polyklonale Population kann mithilfe von Technologien neuartige Next Generation Sequencing (NGS) erreicht werden. Aus diesen Gründen stellen wir Tiefe Sequenzierung Analyse der Bibliothek selbst und die Auswahl-Ausgänge, komplette Informationen über Vielfalt, Fülle und genaue Kartierung aller ausgewählten Fragment. Die hier vorgestellten Protokolle zeigen die wichtigsten Schritte für Bibliotheksbau, Charakterisierung und Validierung.
Einleitung
Hier beschreiben wir eine Hochdurchsatz-Methode für den Bau und die Auswahl der Bibliotheken der gefalteten und lösliches Protein Domänen aus jeder genic/genomische ab Quelle. Der Ansatz kombiniert drei verschiedene Technologien: Phagen-Display, die Verwendung eines klappbaren Reporter und nächsten Generation Sequencing (NGS) mit einer speziellen Web-Tool für die Datenanalyse. Die Methoden können in vielen verschiedenen Zusammenhängen von Protein-basierten Forschung verwendet werden, für Identifikation und Kommentierung der neuen Proteine-Eiweiß-Domains, Charakterisierung von strukturellen und funktionellen Eigenschaften des bekannten Proteine sowie Definition von Protein-Interaktion Netzwerk.
Viele offene Fragen sind nach wie vor in Protein-basierten Forschung und die Entwicklung von Methoden für optimale Protein-Produktion ist eine wichtige Notwendigkeit für mehrere Felder der Untersuchung. Zum Beispiel fehlt trotz der Verfügbarkeit von Tausenden von prokaryotischen und eukaryotischen Genomen1, eine entsprechende Karte von der relativen Proteome mit eine direkte Anmerkung der kodierten Proteine und Peptide für die große Mehrheit der Organismen. Der Katalog der komplette Proteome taucht als ein anspruchsvolles Ziel, einen riesigen Aufwand an Zeit und Ressourcen erfordern. Der Goldstandard für experimentelle Annotation bleibt das Klonen von alle Open Reading Frames (ORFs) eines Genoms Bau der so genannten "ORFeome". In der Regel Genfunktion Basierend auf Homologie zu verwandten Genen bekannte Tätigkeit zugewiesen, aber dieser Ansatz ist schlecht genau aufgrund der Anwesenheit von vielen falschen Anmerkungen in der Referenz Datenbanken2,3,4, 5. Darüber hinaus auch für Proteine, die identifiziert und kommentiert haben, zusätzliche Studien sind verpflichtet, Charakterisierung in Bezug auf Fülle, Expressionsmuster in unterschiedlichen Kontexten, einschließlich der strukturelle und funktionelle Eigenschaften sowie Interaktion-Netzwerke.
Darüber hinaus da Proteine aus verschiedenen Domänen, jeder von ihnen bestehen zeigt Besonderheiten und anders zu Proteinfunktionen beitragen, die Studie und die genaue Definition dieser Domains können ein noch umfassenderes Bild, sowohl bei den einzelnen gen und auf das vollständige Genom-Ebene. Diese notwendigen Informationen macht Protein-basierten Forschung einem breiten und anspruchsvollen Bereich.
In dieser Perspektive könnte ein wichtiger Beitrag durch unvoreingenommene und Hochdurchsatz-Methoden für die Proteinproduktion gegeben werden. Allerdings beruht der Erfolg solcher Ansätze, neben der erheblichen Investition erforderlich ist, auf der Fähigkeit, lösliche/Stall Protein Konstrukte zu produzieren. Dies ist ein wichtiger Faktor zu begrenzen, da es wurde geschätzt, dass nur etwa 30 % der Proteine können erfolgreich ausgedrückt und produziert in ausreichendem Maße experimentell nützlich6,7,8. Eine Annäherung an diese Beschränkung zu überwinden basiert auf der Verwendung von nach dem Zufallsprinzip fragmentierte DNA herstellen verschiedene Polypeptide, die zusammen überlappende Fragment Darstellung einzelner Gene. Nur ein kleiner Prozentsatz der zufällig generierte DNA-Fragmente sind funktionale ORFs, während die große Mehrheit von ihnen sind nicht-funktionale (wegen des Vorhandenseins des Stop-Codons in ihre Sequenzen) oder codieren für unnatürlich (ORF in einem Frame als das Original) Polypeptide mit keine biologische Bedeutung.
Um diese Probleme zu beheben, hat unsere Fraktion eine Hochdurchsatz-Protein Ausdruck und Interaktion Analyse-Plattform entwickelt, die auf genomischer Ebene9,10,11,12verwendet werden kann. Diese Plattform integriert die folgenden Techniken: 1) eine Methode, um Sammlungen von korrekt gefaltete Protein Domains wählen Sie die Codierung Teil der DNA von jedem Organismus; (2) die Phagen-Display-Technologie für die Auswahl der Partner von Interaktionen; (3) die NGS vollständig charakterisieren die ganze Interaktom untersucht und die Klone von Interesse zu identifizieren; und 4) eine Web-Tool für die Datenanalyse für Anwender ohne Programmierkenntnisse oder Bioinformatik Interaktom-Seq-Analyse in eine einfache und benutzerfreundliche Weise durchführen.
Die Nutzung dieser Plattform bietet wichtige Vorteile gegenüber alternativen Strategien der Untersuchung; die Methode ist vor allem völlig unvoreingenommene, Hochdurchsatz- und modular für Studie, die von einem einzigen Gen bis zu einem ganzen Genom. Der erste Schritt der Pipeline ist die Schaffung einer Bibliothek von nach dem Zufallsprinzip fragmentierte DNA unter Studie, die von NGS dann tief geprägt ist. Diese Bibliothek wird über ein veränderter Vektor wo werden Gene/Fragmente des Interesses geklont, zwischen einer Signalsequenz für Protein Sekretion in den periplasmatischen Raum (d.h. ein Sec-Führer) und das TEM1 β-Lactamase-gen erzeugt. Fusionsproteins wird Ampicillin-Resistenz und die Fähigkeit, unter Druck von Ampicillin nur überleben, wenn geklonten Fragmente im Rahmen sind verleihen diese Elemente und die daraus resultierenden Schmelzverfahren Protein ist korrekt gefaltet10,13 ,14. Alle Klone gerettet, nachdem Antibiotika Auswahl, die so genannte "gefiltert-Klone", sind ORFs und eine große Mehrheit von ihnen (mehr als 80 %), echte Gene9abgeleitet sind. Darüber hinaus liegt die Stärke dieser Strategie in die Ergebnisse, dass alle ORF gefiltert Klone für korrekt gefaltet/lösliche Proteine/Domänen15codieren. Wie viele Klone, vorhanden in der Bibliothek und die Zuordnung in der gleichen Region/Domäne, verschiedene Start- und Endpunkte haben, ermöglicht das unvoreingenommene, einstufigen Identifikation der minimale Fragmente, die lösliche Produkte führen dürften.
Eine weitere Verbesserung in der Technologie ist durch die Verwendung von NGS gegeben, um die Bibliothek zu charakterisieren. Die Kombination dieser Plattform und eine spezielle Web-Tool für die Datenanalyse gibt wichtige objektive Informationen über die genauen Nukleotidsequenzen und über den Standort des ausgewählten ORFs auf dem Referenz-DNA unter Studie ohne weitere umfangreiche Analysen oder experimentellen Aufwand.
Domainome Bibliotheken können in einem Auswahlkontext übertragen und als ein universelles Instrument verwendet, um funktionelle Studien durchführen. Die Hochdurchsatz-Protein Ausdruck und Interaktion Analyseplattform, dass wir integriert und dass wir Interaktom-Seq genannt die Phagen-Display-Technologie nutzt durch Übertragung den gefilterten ORF in einen Phagemid Vektor und die Schaffung von einem Phagen-ORF Bibliothek. Einmal neu geklont in einem Phagen Anzeigekontext Protein Domains auf der Oberfläche der M13 Partikel angezeigt; auf diese Weise können Domainome Bibliotheken für Genfragmente Codierung Domains mit spezifischen enzymatischen Aktivitäten oder bindende Eigenschaften, so dass Interaktom Netzwerke Profilerstellung direkt ausgewählt werden. Dieser Ansatz wurde ursprünglich von Zacchi Et Al. beschrieben. 16 und später in mehreren anderen Kontext13,17,18verwendet.
Im Vergleich zu anderen Technologien zur Untersuchung von Protein-Protein-Interaktion (einschließlich Hefe-zwei-Hybrid-System und Massenspektrometrie19,20), ist ein großer Vorteil die Verstärkung der Bindungspartner, die auftritt, während Phagen mehrfache Umläufe der Auswahl angezeigt. Dies erhöht die Auswahl-Empfindlichkeit, so dass die Identifizierung der niedrigen reichlich Bindeproteine Domänen in der Bibliothek vorhanden. Die Effizienz der Auswahl mit ORF-gefilterte Bibliothek durchgeführt wird aufgrund des Fehlens von nicht-funktionalen Klone weiter erhöht. Schließlich ermöglicht die Technologie die Auswahl gegen Eiweiß und nicht-Protein Köder21,22,23,24,25durchgeführt werden.
Phagen-Auswahl mit Hilfe der Domainome-Phagen-Bibliothek können mit Antikörpern im Serum von Patienten mit verschiedenen pathologischen Bedingungen, z.B. Autoimmunerkrankungen13, Krebs oder Infektionen Krankheiten als Köder aus durchgeführt werden. Dieser Ansatz wird verwendet, um zu erhalten die so genannte "Antikörper Signatur" der Krankheit unter Studie damit massiv zu identifizieren und zu charakterisieren die Antigene/Epitope speziell zur gleichen Zeit von der Patienten Antikörper erkannt. Im Vergleich zu anderen Methoden der Verwendung von Phagen-Display ermöglicht die Identifikation von sowohl lineare als auch Konformationsänderungen Antigenen Epitope. Die Identifikation einer bestimmten Signatur könnte einen wichtigen Einfluss für Verständnis Pathogenese, neue Impfstoff-Design, Identifikation von neuen therapeutischen Ziele und Entwicklung von neuen und spezifischen diagnostischen und prognostischen Tools haben. Darüber hinaus Wenn die Studie auf Infektionskrankheiten konzentriert ist, ist ein großer Vorteil, dass die Entdeckung der Immunogenen Proteine unabhängig vom Erreger Anbau.
Unser Ansatz bestätigt, dass die Faltung Reporter auf genomischer Ebene verwendet werden können, um die "Domainome" wählen: eine Sammlung von korrekt gefaltet, gut ausgedrückt, lösliches Protein Domains aus der Codierung Teil der DNA bzw. cDNA von jedem Organismus. Einmal isoliert die PROTEINFRAGMENTE eignen sich für viele Zwecke, mit wesentlichen experimentelle Informationen für gen-Anmerkungen sowie für strukturelle Studien, Antikörper Epitop Mapping, Antigen-Identifikation. Die Vollständigkeit der Hochdurchsatz-Daten zur Verfügung gestellt von NGS ermöglicht die Analyse von hochkomplexen Proben, wie Phagen-Display-Bibliotheken, und hat das Potenzial, das traditionelle mühsam sammeln und Prüfung von einzelnen Phagen gerettet Klone zu umgehen.
Zur gleichen Zeit dank der Funktionen der gefilterten Bibliothek und die extreme Empfindlichkeit und macht die NGS-Analyse ist es möglich, die Protein-Domäne verantwortlich für jede Interaktion direkt in ein Einstiegsbild, ohne die Notwendigkeit der Schaffung zu identifizieren zusätzliche Bibliotheken für jede gebunden Protein. NGS ermöglicht eine umfassende Definition von der ganzen Domainome gen/genomische ab Quelle und das Web-Tool zur Datenanalyse ermöglicht die Erlangung von eine hochspezifische Charakterisierung von qualitativer und quantitativer Sicht von der Interaktom Proteine Domänen.
Protokoll
1. Aufbau der ORF-Bibliothek (Abbildung 1)
- Vorbereitung der Einsatz DNA
-
Vorbereitung von synthetischen oder genomische DNA-Fragmente
- Extrakt/DNA mit Standardmethoden26zu reinigen.
- Fragment-DNA durch Ultraschallbehandlung. Wenn mit einem standard sonikator als allgemeine Anregung Start mit 30 s-Pulse bei 100 % Leistung.
Hinweis: Pilot Versuche sollten mit unterschiedlicher Leistung und Beschallung Zeiten legen Sie die optimalen Bedingungen für die DNA-Vorbereitung durchgeführt werden. Bestimmen Sie nach jedem Test die Größe der DNA-Fragmente durch Agarose-Gelelektrophorese. - Laden Sie die beschallte DNA auf 1,5 % Agarosegel, zusammen mit einer 100 bp DNA-Leiter. Führen Sie eine kurze Elektrophorese bei 5 V/cm für 15 min laufen und schneiden Sie den Teil des Gels mit dem Abstrich der fragmentierten DNA.
- Reinigen Sie die Einsatz DNA mit einem spaltenbasierten Gel Extraction Kit und Messen Sie die Konzentration mit einer UV-Spektralphotometer.
Hinweis: mindestens 500 ng von gereinigten Beilagen erhalten Sie nach diesem Schritt, um mit 1 µg der verdauten Vektor ligiert werden, wie unter Punkt 1.3 beschrieben. Prüfen Sie die Qualität der Fragmente Vorbereitung durch die Auswertung eine260nm/a280nm und ein260nm/a230nm Verhältnisse, da niedriger Qualität der Probe die Ligatur Effizienz auswirkt. - Bis zu 5 μg der Einsätze mit 1 μl Quick Abstumpfung Kit-Enzym-Mix, laut Angaben des Herstellers zu behandeln. Inaktivieren Sie Enzyme durch Erhitzen auf 70 ° C für 10 min. Proben bis zur Verwendung bei-20 ° C gelagert werden können.
-
Vorbereitung von cDNA-Fragmente
- Extrahieren Sie RNA mit Standardmethoden (z. B. mit TRizol oder ähnlichen Reagenzien).
- Fragment-mRNA durch Erhitzen vor dem Durchführen der reversen Transkription. Die endgültige Länge der DNA-Fragment wird von mRNA kochende Zeit und zufällige Grundierung Konzentration gesteuert. Zum Beispiel Wärme Probe für 6 min bei 95 ° C.
- Bereiten Sie cDNA mit zufälligen Primern mit jeder verfügbaren Kit nach Protokoll des Herstellers vor.
- Zum Abbau cDNA von Poly-dT Tails durch Hybridisierung mit biotinylierte Poly-dA für 3 h bei 37 ° C und auf Streptavidin magnetische Beads zu trennen, wie von Carninci Et Al. beschrieben 13
- Ungebundenes Material zu erholen und zu reinigen mit einem spaltenbasierten DNA Reinigung Kit nach den Anweisungen des Herstellers. Messen Sie die Konzentration, die mit einem UV-Spektralphotometer. Siehe Hinweis im Schritt 1.1.1.4..
-
Vorbereitung von synthetischen oder genomische DNA-Fragmente
- Vorbereitung der Filterung Vektor
- Digest 5 µg des gereinigten klonen Vektor pFILTER312 mit 10 U EcoRV Restriktionsenzym, folgende Hersteller-Protokoll.
- Laden 2 µL (200 ng) der verdauten Vektor, zusammen mit 100 ng des unverdauten Vektors und 1 k bp Molekulare Marker, auf einem 1 % Agarosegel, um für die richtige Verdauung zu überprüfen. Wärme zu inaktivieren das Restriktionsenzym.
- Volumen 1/10 10 x Phosphatase Puffer und 1 µL (5 U) Phosphatase und Inkubation bei 37 ° C für 15 min. Hitze zu inaktivieren, für 5 min bei 65 ° C.
- Verdaute Plasmid durch Extraktion aus Agarosegel zu reinigen, und die Konzentration mit einer UV-Spektralphotometer messen. Proben können bis zur Verwendung bei-20 ° C gelagert werden.
- Ligatur und transformation
- Ligatur wie folgt durchführen: 1 µg der verdauten Plasmid hinzufügen 400 ng der phosphorylierten Einsätze (Plasmid: Insert Molverhältnis 1:5), 10 µL 10-fach Puffer für T4 DNA-Ligase, 2 µL hohe Konzentration T4 DNA-Ligase in einem Endvolumen von 100 µL. brüten die Reaktion bei 16 ° C Leid GHT. Wärme bei 65 ° C für 10 min zu inaktivieren.
- Überstürzen sich die Ligatur Produkt durch Zugabe von 1/10-bändige Natrium-Acetat-Lösung (3 M, pH 5,2) und 2,5 Volumina von 100 % Ethanol. Mischen und bei-80 ° C für 20 min einfrieren.
- Zentrifugieren Sie mit maximaler Geschwindigkeit für 20 min bei 4 ° C. Verwerfen Sie den überstand.
- Pellet und Zentrifuge mit maximaler Geschwindigkeit für 20 min bei 4 ° c kaltem 70 % igem Ethanol 500 µL hinzufügen Verwerfen Sie den überstand.
- Das Pellet der Luft trocknen lassen. Die ausgefällte DNA in 10 µL Wasser aufschwemmen.
- Durchführen Sie Bakterienzelle Elektroporation.
Hinweis: Die Verwendung von hocheffizienten Zellen (über 5 x 109 transformanten pro µg DNA) ist erforderlich. Wir empfehlen die Verwendung von Escherichia coli DH5αF "(F'/ endA1 hsd17 (rK − mK +) supE44 Thi-1recA1 GyrA (Nalr) relA1 (LacZYA-ArgF) U169 DeoR (F80dlacD-(lacZ)M15) produziert im eigenen Haus oder von mehreren Herstellern gekauft.- Legen Sie entsprechende Anzahl von Mikrozentrifugenröhrchen und 0,1 cm-Elektroporation Küvetten auf Eis. 25 µL der Zellen 1 µL der gereinigten Ligatur-Lösung (in VE-Wasser) hinzufügen und flick das Rohr ein paar Mal.
- Übertragen der DNA-Zellen-Mischung auf die kalten Küvette, tippen Sie auf Arbeitsplatte 2 X, wischen Sie Wasser von außen der Küvette, legen Sie in die Elektroporation Modul und Presse Puls.
- Führen Sie Elektroporation mit einer standard Electroporator-Maschine mit 25 µF, 200 Ω und 1,8 kV. Zeitkonstante muss 4 bis 5 ms liegen.
- Sofort 1 mL der flüssigen 2xYT Medium ohne jedes Antibiotikum, übertragen auf eine 10 mL-Tube und lassen bei 37 ° C, schütteln bei 220 u/min für 1 h wachsen.
- Platte verwandelt DH5αF' auf 15 cm 2xYT Agarplatten mit 34 µg/mL Chloramphenicol (pFILTER Widerstand) und 25 µg/mL Ampicillin (selektive Marker für ORFs) ergänzt und Inkubation über Nacht bei 30 ° C.
- Platte Verdünnungen der Bibliothek auf 10 cm 2xYT Agarplatten ergänzt mit Chloramphenicol + Ampicillin und Chloramphenicol nur, Bibliothek Titration durchführen. Inkubation über Nacht bei 30 ° C.
- pFILTER-ORF-Bibliothek-Validierung
- Testen Sie 15-20 Kolonien von Chloramphenicol und Chloramphenicol/Ampicillin Platten, Insert Größenverteilung zu schätzen. Wählen Sie einzelne Kolonien mit einer Spitze und verdünnen sie separat in 100 µL des 2xYT Mediums ohne Antibiotika. Verwenden Sie 0,5 µL dieser Lösung als DNA-Vorlage für eine PCR-Reaktion mit jedem standard TaqDNA-Polymerase nach Protokoll des Herstellers.
- Führen Sie 25 Zyklen der Amplifikation mit einem T Glühen von 55 ° C und eine Verlängerung von 40 s bei 72 ° C. Primer-Sequenzen sind in der Tabelle der Materialienzur Verfügung gestellt.
- Laden Sie die PCR-Produkte auf 1,5 % Agarosegel, zusammen mit einer 100 bp DNA-Leiter und Lauf.
- pFILTER-ORF-Bibliothek
- Sammeln Sie Bakterien aus den 150 mm Platten durch Zugabe von 3 mL frische 2xYT Medium zu und ernten sie mit einem sterilen Schaber, gründlich mischen, mit 20 % steriles Glyzerin zu ergänzen und bei-80 ° C in kleinen Aliquote lagern.
- Reinigen von Plasmid-DNA aus einem aliquoten der Bibliothek (vor der Zugabe von Glycerin) mit einem spaltenbasierten Plasmid Extraktion Kit, nach Anweisungen des Herstellers.
- Messen Sie die Konzentration mit der UV-Spektralphotometer. Proben können gespeichert werden, bei-20 ° C bis zur Phagemid Bibliothek Vorbereitung bzw. Charakterisierung von NGS verwendet werden.
(2) subcloning der gefilterten ORFs in einem Phagemid-Vektor (Abbildung 2)
-
Vorbereitung des ORF gefiltert DNA-Fragmente
- Beschränkung Enzym Verdauung von 5 µg des gereinigten Vektor aus dem pFILTER-ORF-Bibliothek-Vektor hinzufügen 10 U von BssHII und Inkubation entsprechend des Herstellers Protokoll eingerichtet. Inaktivieren Sie das Enzym und Digest mit 10 U NheI.
- Laden Sie die verdaute DNA auf 1,5 % Agarosegel, zusammen mit einer 100 bp DNA-Leiter. Führen Sie eine kurze Elektrophorese laufen bei 5 V/cm für 15 min oder gerade genug um den Abstrich der ausgeschnittenen Fragmente zu unterscheiden und schneiden Sie den Teil des Gels, die sie enthalten.
- Reinigen Sie die Einsatz DNA mit einem Spalte-Base Gel Extraction Kit und Messen Sie die Konzentration mit einer UV-Spektralphotometer.
-
Vorbereitung der Phagemid DNA
- Richten Sie Beschränkung Enzym Verdauung von 5 µg des gereinigten pDAN527 für die Einsätze.
- Verdaute Plasmid durch laufende verdaute DNA auf einem 0,75 % Agarose-Gel reinigen und Auszug aus dem Gel mit einem spaltenbasierten Kit.
- Messen Sie die Konzentration, die mit einem UV-Spektralphotometer. Proben können bis zur Verwendung bei-20 ° C gelagert werden.
-
Bibliothek-Ligatur, Transformation und Sammlung
- Durchführen Sie Ligatur und Transformation für pFILTER Vektor beschrieben.
- Platte verwandelt DH5αF "auf 150 mm 2xYT Agarplatten mit 100 µg/mL Ampicillin ergänzt und Inkubation über Nacht bei 30 ° C.
- Platte Verdünnungen der Bibliothek auf 100 mm 2xYT Agarplatten mit 100 µg/mL Ampicillin zum Bestimmen der bibliotheksgröße ergänzt.
- Durchführen Sie Bibliothek-Überprüfung mittels PCR zufällig ausgewählten Klone, wie unter Punkt 1.4 beschrieben.
- Phagemid-ORF-Bibliothek durch das Ernten von Bakterien aus 150 mm Platten zu sammeln, mischen, mit 20 % steriles Glyzerin und Store bei-80 ° C in kleinen Aliquote ergänzen.
- Reinigen Sie Plasmid DNA aus einer Teilprobe der Bibliothek mit einem spaltenbasierten Plasmid-Extraktion-Kit nach Herstellerangaben.
- Messen Sie die Konzentration auf die UV-Spektralphotometer. Proben können gespeichert werden, bei-20 ° C bis zur Charakterisierung von NGS verwendet werden.
(3) Phagen Bibliothek Vorbereitung und Auswahlverfahren
- Phagen-Produktion
- Verdünnen Sie ein Lager Aliquot der Phagemid Bibliothek in 10 mL 2xYT flüssige Brühe mit 100 µg/mL Ampicillin ergänzt, um eine OD600nm haben = 0,05.
- Wachsen die verdünnte Bibliothek in ein steriles Auffanggefäß 5-10 mal größer als das ursprüngliche Volume, bei 37 ° C mit schütteln bei 220 u/min bis erreicht600nm OD = 0,5.
- Infizieren Sie Bakterien mit Helfer Phagen (z.B. M13K07) bei einer Vielzahl von Infektionen 20:1. Lassen Sie bei 37 ° C für 45 Minuten unter gelegentlichen rühren (alle 10 min).
- Zentrifugieren Sie Bakterien bei 4000 X g für 10 min bei Raumtemperatur. Überstand verwerfen, neu aussetzen Bakterien Pellet in 40 mL flüssige Brühe 2xYT ergänzt mit 100 µg/mL Ampicillin und 50 µg/mL Kanamycin und wachsen bei 28 ° C mit schütteln bei 220 u/min für eine Nacht.
- Am Tag danach, Zentrifuge Bakterien bei 4000 X g für 20 min bei 4 ° C. Den Überstand mit Phagen zu sammeln.
- PEG-Fällung von Phagen.
- Fügen Sie 1/5 Volumen von 0,22 µm PEG/NaCl-Lösung (w 20 % / PEG 6000 V 2,5 M NaCl) auf den geräumten Phagen gefiltert und inkubieren Sie für 30-60 min auf Eis.
Hinweis: Lösung wurde rauchigen nach wenigen Minuten einen erfolgreiche Phagen Niederschlag angibt. Die Trübung der Lösung erhöht über die Inkubationszeit. - Zentrifuge bei 4000 X g für 15 min bei 4 ° C. Einen weißen kleinen Pellets von Phagen bilden.
- Aufschwemmen Sie es in 1 mL sterile PBS. Transfer zum 1,5 mL-Tube und Zentrifugieren bei 4 ° C für 10 min bei maximaler Geschwindigkeit, Verunreinigung Bakterien zu entfernen. Eine braune Kugel bilden.
- Übertragen Sie überstand mit Phagen auf einen neuen Schlauch. Halten Sie Phagen auf Eis für aufeinander folgende Titration und Phagen-Auswahl.
- Fügen Sie 1/5 Volumen von 0,22 µm PEG/NaCl-Lösung (w 20 % / PEG 6000 V 2,5 M NaCl) auf den geräumten Phagen gefiltert und inkubieren Sie für 30-60 min auf Eis.
- Phagen-titration
- Bereiten Sie serielle Verdünnungen der Phagen-Lösung. Setzen Sie 10 µL der Phagen-Lösung in 990 µL PBS 10-2 Verdünnung zu erhalten. Dieses Präparat 10-4 zu machen und daraus erhalten eine 10-6 Verdünnung wieder zu verdünnen.
- DH5αF wachsen "Bakterienzellen in 2xYT flüssigen Medium bei 37 ° C mit schütteln bis OD600nm = 0,5 erreicht ist. 1 mL der vorbereiteten Bakterien in 1,5 mL-Tube übertragen und sofort mit 1 µL 10-4 Phagen Verdünnung zu infizieren. Ohne schütteln bei 37 ° C für 45 min. Wiederholen Sie das gleiche Verfahren für die 10-6 Verdünnung inkubieren.
- Platte Verdünnungen der infizierten Bakterien in 100 mm 2xYT Platte. Legen Sie die Platte über Nacht bei 30 ° C.
- Platte 100 µL nicht infizierten DH5αF "auf einer 2xYT-Agar-Platte ergänzt mit 100 µg/mL Ampicillin, das Fehlen einer Kontamination bei der Vorbereitung zu überprüfen.
- Am Tag nach die Anzahl der Kolonien und der Phage Titer zu berechnen. Express-Titer als Anzahl von Bakterien/mL. Erwarteten Titer ist 10-12-13 -Phagen/mL.
- Phagen-Selektion
-
Phagen-Selektion mit als Köder gereinigt Antikörper
- Sättigen Sie Phagen durch Verdünnung 200 µL des Phagen Vorbereitung in ein gleiches Volumen PBS - 4 % Magermilch zu und inkubieren Sie für 1 h bei Raumtemperatur in langsame Rotation. Dieser Schritt ermöglicht das Blockieren von Phagen für unspezifische Bindung. Übertragen Sie 30 µL Protein-G beschichtete magnetische Beads auf einen 1,5 mL-Tube.
- Zwei Mal wie folgt waschen: Hinzufügen 500 µL PBS, auf einem Rad in langsame Rotation für 2 min bei Raumtemperatur inkubieren, zeichnen Sie die Perlen auf der einen Seite des Rohres mit einem Magneten und den überstand zu entfernen.
- Inkubieren Sie gesättigte Phagen mit gewaschenen Perlen für 30 min bei Raumtemperatur bei langsamer Drehung.
- Zeichnen Sie die Perlen auf der einen Seite mit einem Magnetfeld. Sammeln des Überstands mit Phagen für Auswahlschritt verwendet werden.
- Bereiten Sie magnetische Beads während der Durchführung der vorherigen Schritt durch die gereinigte Antikörper Konjugation vor. Waschen Sie 30 µL Protein-G beschichtete magnetische Beads, wie oben beschrieben. 10 µg gereinigte Antikörper in 500 µL PBS verdünnen, die gewaschenen Perlen hinzu und brüten in langsame Rotation bei Raumtemperatur für 45 min. zweimal mit PBS waschen.
Hinweis: Führen Sie zwei verschiedene Zubereitungen von magnetischen Beads: eins mit Antikörpern von Interesse und eins mit Kontrolle Antikörper, z. B. Antikörper von gesunden Spendern gereinigt. Die Reihenfolge der Antigene mit Kontrolle, die Antikörper während der Analyseschritt der Ausgänge abgezogen werden ausgewählt. Alternativ können magnetische Beads mit Kontrolle Antikörper beladen, einen Pre-Clearing-Schritt von den Phagen durchzuführen (Folgen das Protokoll für die Inkubation mit UN-konjugierten Perlen) verwendet werden. - Phagen-Selektion: Zeichnen Perlen auf der einen Seite des Rohres mit einem Magneten, die letzten Wäsche zu entfernen, hinzufügen Phagen und inkubieren Sie bei langsamer Drehung bei Raumtemperatur für 90 min. 5 Mal mit 500 µL PBS-0.1% Tween-20 und 5 Mal mit PBS waschen.
- Eluieren gebundene Phagen, repräsentieren die Ausgabe der Auswahl durch die Vermischung der Perlen mit 1 mL DH5αF' Zellen gezüchtet bei OD600 = 0,5. Inkubieren Sie Bakterien mit Perlen für 45 min bei 37 ° C mit gelegentlichen schütteln (alle 10 min). Platte der Ausgabe auf einem 150 mm 2xYT Agarplatte mit 100 µg/mL Ampicillin ergänzt.
- Platte 100 µL unverdünnte und verschiedene Verdünnungen des Ausgangs (10-1 bis 10-5) um Titration durchzuführen. Am Tag nach Bakterien von den 150 mm Platten durch Zugabe von 3 mL frische 2xYT Medium zu sammeln und ernten sie mit einem sterilen Schaber gründlich mischen, mit 20 % steriles Glyzerin zu ergänzen und bei-80 ° C in kleinen Aliquote lagern.
- Eine Aliquote erneut aus, um eine Sekunde durchführen Runde Auswahl zu wachsen. Wiederholen Sie das panning Verfahren, wie oben beschrieben, abgesehen von der Wasch-Bedingungen. In diesem Fall 10 mal waschen mit PBS - 1 % Tween-20 (Lösung in den Schlauch Gießen und Gießen Sie sofort wieder). Dann fügen Sie 500 µL PBS und brüten auf Rotation bei Raumtemperatur für 10 min. führen andere 10 Wäschen mit PBS. Fahren Sie mit der Elution Schritt für die erste Runde der Auswahl.
- Ein Aliquot der Ausgabe mit einem spaltenbasierten Kit nach Herstellerangaben extrahieren Sie Plasmid DNA. Speichern Sie Plasmid bei-20 ° C, bis es für tiefe Sequenzierung verwendet wird.
-
Verwendung als Köder rekombinante Proteine Phagen-Selektion
- Sättigen Sie Phagen durch Verdünnung 200 µL des Phagen Vorbereitung in ein gleiches Volumen PBS - 4 % Magermilch zu und inkubieren Sie für 1 h bei Raumtemperatur in langsame Rotation.
- Fügen Sie 100 µL Streptavidin magnetischen Kügelchen. Inkubation für 1 h bei Raumtemperatur Streptavidin-Bindung Phagen auswählen. Streptavidin-gebundenen Phagen zu entfernen, indem man die Perlen auf der einen Seite mit einem Magneten. Nehmen Sie überstand aus dem vorherigen Schritt und fügen Sie biotinylierte Protein (in einer Konzentration von 100-550 nM) und auf ein Rotor für 30 min bis 1 h bei Raumtemperatur inkubieren.
- Bereiten Sie magnetische Beads: während der Durchführung der vorherigen Schritt, 100 µL Streptavidin-magnetische Beads mit PBS waschen in PBS 2 % Magermilch aufschwemmen und inkubieren mit Rotation bei Raumtemperatur für 30 min bis 1 h.
- Phagen-Selektion: Perlen auf der einen Seite des Rohres mit einem Magneten zu zeichnen, PBS - 2 % Milch zu entfernen und Aufschwemmen Perlen mit Phagen-Protein-Mix. Bei langsamer Drehung bei Raumtemperatur für 90 min inkubieren.
- Perlen auf der einen Seite des Rohres mit einem Magneten zu zeichnen, den überstand verwerfen und waschen Sie sie sorgfältig fünfmal mit 500 µL PBS 0,1 % Tween-20. Führen Sie Elution, wie in der vorherigen Sitzung beschrieben.
-
Phagen-Selektion mit als Köder gereinigt Antikörper
(4) Phagen Bibliothek Deep Sequencing Plattform (Abbildung 3)
-
DNA fügt Wiederherstellung nach pFILTER-ORF-Bibliothek, pDAN5-ORF-Bibliothek oder ausgewählt-Phagen-Bibliotheken
- Tauwetter ein Aliquot der Bibliothek, mit einem Spektrophotometer zu quantifizieren, DNA-Einsätze durch Amplifikation mit spezifischen Zündkapseln zu erholen.
Hinweis: Die Primer verwendet, um die Einsätze zu retten hängen an ihrem 5'-Ende Adapter Sequenzen, so dass die aufeinander folgenden Indizierung der Amplifikate Pools erhalten und die direkte Sequenzierung der DNA-Einsätze mit dem Sequenzer wiederhergestellt. Deren Reihenfolge ist in der Tabelle der Materialien. Die Adapter sind in Fettdruck, angegeben und die spezifische Primer sind in Kursivschrift angegeben. - Verwenden Sie 2,5 µL der Bibliothek (pFILTER/Phagemid/ausgewählt-Phagen) als DNA-Vorlage für eine PCR-Reaktion.
- Verwenden Sie das folgende Programm: 95 ° C für 3 min; 25 Zyklen von 95 ° C für 30 s, 55 ° C für 30 s, 72 ° C für 30 s; 72 ° C für 5 min. Halt bei 4 ° C.
Hinweis: an dieser Stelle empfiehlt es laufen 1 µL des PCR-Produkts auf einem Bioanalyzer oder TapeStation, überprüfen die Größe der Amplifikate und überprüfen sie in den richtigen Bereich.
- Tauwetter ein Aliquot der Bibliothek, mit einem Spektrophotometer zu quantifizieren, DNA-Einsätze durch Amplifikation mit spezifischen Zündkapseln zu erholen.
-
PCR-Bereinigung
- Bringen Sie die magnetische Beads (z. B. AMPure) auf Raumtemperatur. Übertragen Sie das gesamte PCR-Produkt aus dem PCR-Röhrchen auf eine 1,5 mL-Tube. Wirbel der magnetischen Beads für 30 s um sicherzustellen, dass die Perlen gleichmäßig verteilt sind. Jedes Rohr das PCR-Produkt, Mix von sanft Pipettieren mit fügen Sie 20 µL des magnetischen Beads hinzu. Bei Raumtemperatur ohne Schütteln für 5 min inkubieren.
- Platzieren Sie die Platte auf einem Magnetstativ für 2 min oder bis der Überstand abgeklungen. Mit der PCR-Produkte auf die Magnetstativ entfernen und entsorgen des Überstands.
- Waschen Sie die Perlen mit frisch zubereiteten 80 % Ethanol, mit der PCR-Produkte auf dem magnetischen Stand wie folgt: Fügen Sie 200 µL von frisch zubereiteten 80 % Ethanol zu jeder Probe gut; inkubieren Sie die Platte auf die Magnetstativ für 3 s; sorgfältig entfernen und entsorgen des Überstands.
- Führen Sie eine zweite Ethanol waschen, mit der PCR-Produkte auf dem magnetischen Stand; am Ende des zweiten waschen sorgfältig entfernen Sie das Ethanol und die Perlen für 10 Minuten an der Luft trocknen.
- Entfernen Sie die PCR-Produkte aus der Magnetstativ, jedes Rohr 17,5 µL 10 mM Tris pH 8,5 hinzufügen, pipette vorsichtig oben und unten 10 Mal darauf achten, dass Perlen voll Nukleinsäuretablette sind. In 2 min bei Raumtemperatur inkubieren.
- Setzen Sie das Rohr auf die magnetische Halterung für 2 min oder bis der Überstand geklärt hat, übertragen Sie sorgfältig 15 µL des Überstands mit den gereinigten PCR-Produkten zu einem neuen 1,5 mL-Tube. Speichern Sie die gereinigten PCR-Produkte bei-15 ° C bis-25 ° C bis zu einer Woche, wenn Sie nicht sofort zum Index PCR gehen zu tun.
-
Index-PCR
Hinweis: Nach der PCR bereinigen, durchführen Sie Index PCR. Verwenden Sie das Nextera XT Index-Kit; So wird die daraus resultierende doppelte indizierten Bibliotheken innerhalb gemultiplexten Illumina läuft Sequenz möglich.- Übertragen Sie alle der 15 μl, enthält jedes Produkt zu einem neuen PCR-Schlauch gereinigt und die folgende Reaktion mit einrichten: 15 µL der gereinigten Amplikons Produkt, 5 µL des Index Primer 1 und 5 µL des Index Primer 2, 25 µL 2 X PCR-Mix; Endvolumen von 50 µL.
- Führen Sie PCR auf einem Thermocycler mit dem folgenden Programm: 95 ° C für 3 min, 8 Zyklen von 95 ° C für 30 s, 55 ° C für 30 s, 72 ° C für 30 s; 72° C für 5 min, dann halten Sie bei 4 ° C.
-
PCR Aufräumen 2
- Befolgen Sie das gleiche Protokoll beschrieben im Abschnitt 4.2 für PCR Aufräumen mit folgenden Änderungen: im ersten Schritt 56 µL des magnetischen Beads zu jeweils 50 µL des PCR-Produkt hinzufügen.
- Die Perlen in 27,5 µL 10 mM Tris pH 8,5 im letzten Schritt der Reinigung aufschwemmen und 25 µL zu einem neuen Schlauch (Dies ist die gereinigte letzte Bibliothek bereit zur Quantifizierung und dann Sequenzierung) übertragen.
- Speichern Sie die Platte bei-15 ° C bis-25 ° C bis zu einer Woche, wenn nicht zur Quantifizierung der Bibliothek voran.
-
Qualitative und quantitative Auswertung der Sequenzierung Bibliothek
- Nach der Reinigung, laufen 1 µL einer 01:10 Verdünnung der endgültigen Bibliothek auf einem Bioanalyzer, überprüfen die Größe und Auswahl des für die endgültige Bibliothek Ablaufverfolgung quantifizieren.
- Parallel durchführen Sie die Bibliothek Quantifizierung mittels Real-Time PCR mit einer Bibliothek Quantifizierung Kit pro Protokoll des Herstellers.
-
Bibliotheken, die Sequenzierung
- Die dual indizierten Bibliotheken produzierte zusammen mit anderen Bibliotheken dual indizierten Sequenzierung zu bündeln. Sequenz, die diese Art der Bibliothek durch die Generierung von lange liest, mindestens 250 bp gekoppelten Ende mithilfe der Hiseq2500 oder die MiSeq Instrumente erhalten in den ersten Fall 250bp PE liest und im zweiten Fall 300 bp PE liest.
5. bioinformatische Analyse mithilfe des Interaktom-Seq-Web-Tools
- Analysieren Sie die liest stammt aus pFILTER/Phagemid/ausgewählt-Phagen Bibliothek Sequenzierung mit der Interaktom-Seq-Daten-Analyse-Pipeline. Das Web-Tool ist frei verfügbar unter der folgenden Adresse: http://interactomeseq.ba.itb.cnr.it/
Ergebnisse
Die Filterung Ansatz ist in Abbildung 1schematisiert. Jede Art von intronless DNA kann verwendet werden. In Figur 1A ist der erste Teil der Filterung Ansatz vertreten: nach dem Laden auf einem Agarosegel oder einem Bioanalyzer eine gute Fragmentierung der DNA von Interesse erscheint als ein Abstrich der Fragmente mit einer Länge in der gewünschten Größe 150-750 bp. Eine repräsentative virtuelle Gelbild der fragmentierten DNA gewonnen wird gegeben. Fragmente auf das Agarosegel geladen werden dann wiederhergestellt, Ende repariert phosphoryliert und dann in einen zuvor abgestumpften pFILTER Vektor erstellen Sie eine Bibliothek von zufälligen DNA-Fragmente kloniert. Jedem Schritt des Verfahrens Klonen unter optimalen Bedingungen ist erforderlich, um gute Qualität Bibliothek mit Gesamtabdeckung der DNA unter Studie zu erhalten.
In Abbildung 1 b ist die Filterung Ansatz vertreten: die Bibliothek wächst im Beisein von Chloramphenicol (pFILTER Widerstand) allein oder Chloramphenicol und Ampicillin für ORF-haltigen Kolonien auswählen. Nur Kolonien mit ein DNA-Fragment, ein ORF entspricht eine funktionale β-Lactamase produzieren und überleben, wenn Antibiotika Auswahl vorhanden ist. Abbildung 1 zeigt, wie die Erhöhung Selektionsdruck Auswahl an guten Ordner ORFs gegenüber armen Ordner diejenigen ermöglicht. Das erwartete Ergebnis ist eine Abnahme der Größe der Bibliothek von etwa 20-fold. Höhere Anzahl der Überlebenden Klone zeigt unzureichend Selektionsdruck.
ORF-Fragmente können einfach aus der gefilterten Bibliothek für Folgeantrag wiederhergestellt werden; Unsere Strategie nutzt für Wechselwirkungsstudien Phagen-Display-Technologie. In Abbildung 2sind die wichtigsten Schritte des Phagen Bibliotheksbau vertreten: eine adäquate Bibliothek wird vorbereitet, indem gefilterte Fragmente aus dem pFILTER-Vektor Ausschneiden und wieder Klonen in einem Phagemid Plasmid in Verschmelzung mit der Sequenz-Codierung für die Phagen Capside Protein g3p. Einmal mit Helfer Phagen infiziert, ermöglicht die Anwesenheit des Vektors in Bakterienzellen die Herstellung von Phagen Partikel ORF-g3p Fusion Produkten auf ihrer Oberfläche, wodurch die gefilterte Bibliothek Phagen-Display ausgewählt und weitere anzeigen Analyse.
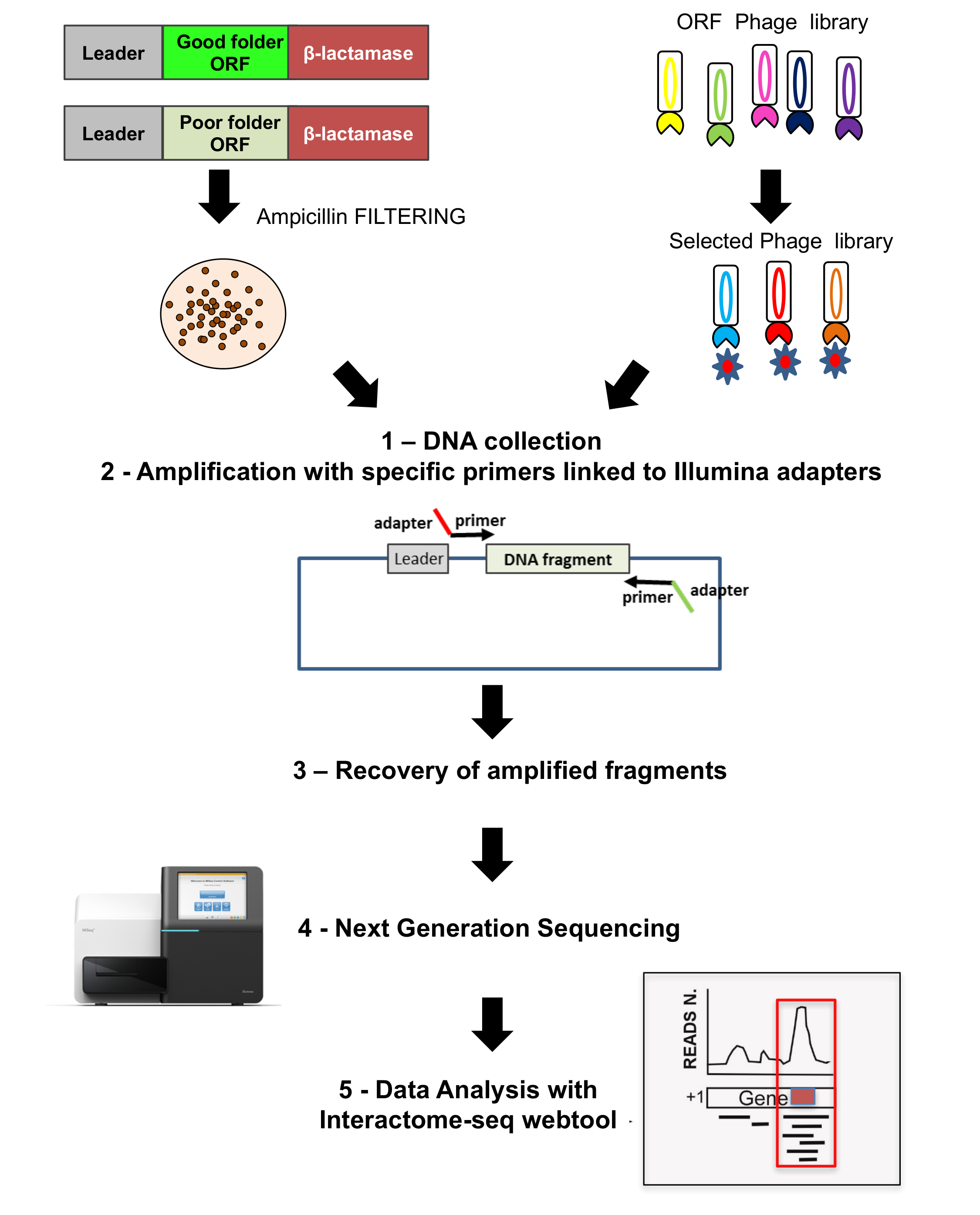
Alle Bibliotheken sind tief von NGS, sowie die Ausgänge der Phagen Auswahlen, analysiert, wie im zweiten Teil von Abbildung 3dargestellt. DNA-Fragmente werden von der wachsenden Kolonien gerettet durch PCR-Amplifikation mit spezifischen Oligonukleotiden glühen auf dem Plasmid-Backbone und Durchführung von speziellen Adaptern für die Sequenzierung. NGS erfolgt und liest dann die Interaktom-Seq Datenanalyse Web-Tool analysiert werden.
In Abbildung 4 berichteten wir eine schematische Darstellung des Auswahlverfahrens einer ORF gefilterte Phagen-Display-Bibliothek. Die Auswahl in diesem Beispiel erfolgt mithilfe von Antikörpern in den Seren von Patienten mit verschiedenen Pathologien (d.h. infektiöse Erkrankungen, Autoimmun-Erkrankungen, Krebs) vorhanden. In diesem Fall interagiert die Phagen-Bibliothek direkt mit den Antikörpern in Seren der Patienten und auf diese Weise wird die vermeintliche spezifische Antigene bereichert werden können, da sie durch Krankheit spezifischen Antikörper erkannt werden. In dieser Art von Experiment ist in der Regel die Bibliothek auch ausgewählt mit Kontrolle Seren von gesunden Patienten um ein Hintergrund-Signal für aufeinander folgende Vergleich und Normalisierung Verfahren verwendet werden.
Auswahl erfolgt über Seren von der gleichen Art von Patienten in der Regel in verschiedenen Pools gruppiert um interindividuelle Variabilität der Sera Antikörper-Titer zu reduzieren. Jeder Pool dient unabhängig für zwei bis drei aufeinander folgenden Runden Auswahl, bereichern die Bibliothek für immun-reaktive Klone, die spezifisch für die Pathologie untersucht. Test Set Antikörper sind mit Bibliothek Phagen inkubiert, immun-komplexe werden von Protein A beschichtet Magnetics-Perlen wiederhergestellt und gebundene Phagen sind durch Standardverfahren eluiert. Die Auswahl-Zyklen werden mit zunehmender waschen und Stringenz verbindlich durchgeführt.
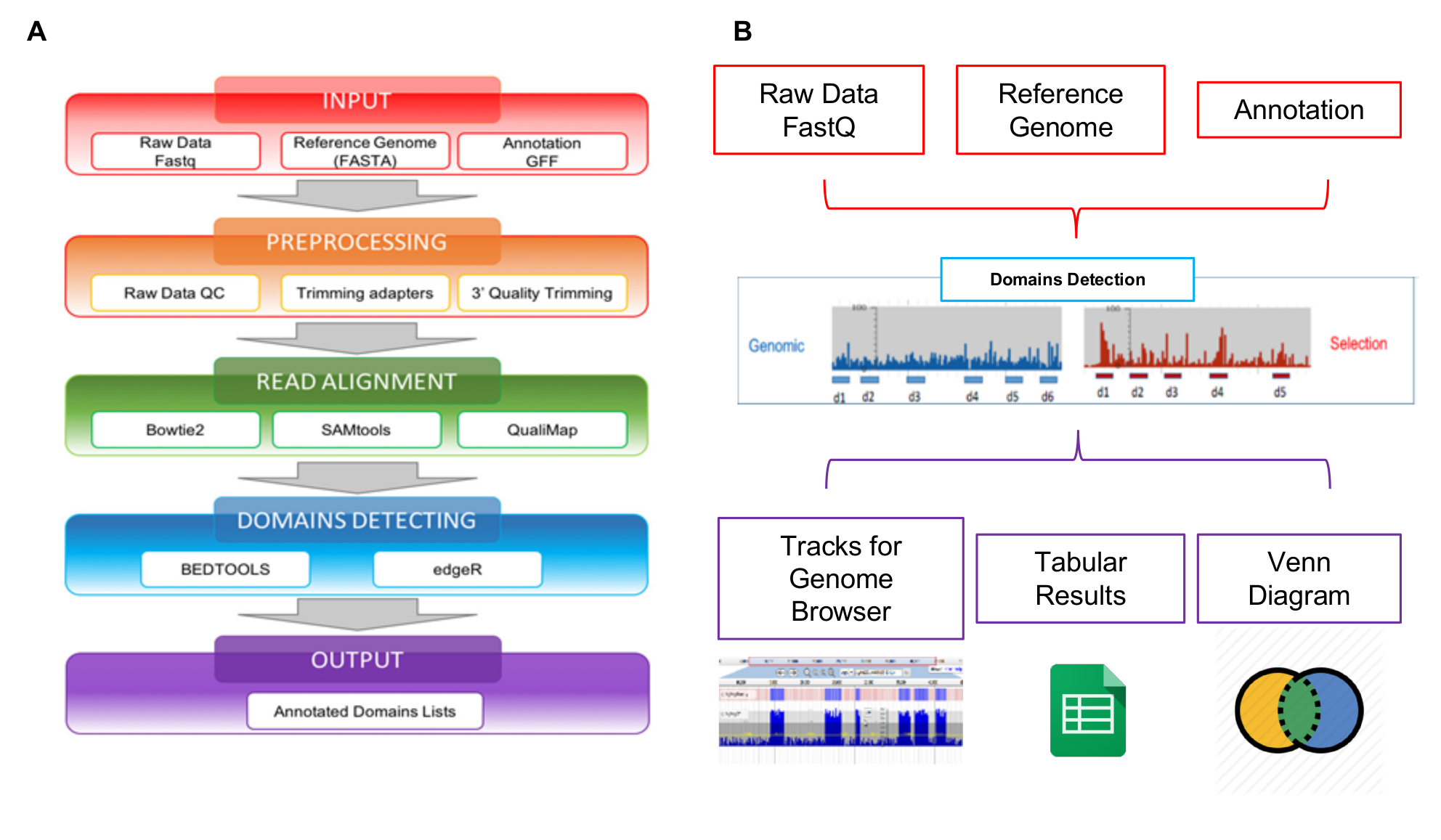
Die Lesevorgänge von NGS erzeugt können das Interaktom-Seq-Web-Tool speziell entwickelt, um diese Art von Daten zu verwalten mit analysiert werden. Interaktom-Seq-Daten-Analyse-Workflow besteht aus vier aufeinander folgenden Schritten, die, ausgehend von rohen Sequenzierung liest die Liste der vermeintlichen Domänen mit genomischen Anmerkungen (Abb. 5A) erzeugt. Im ersten Schritt Eingabe (Abbildung 5A - rotes Kästchen) prüft Interaktom-Seq, ob die input-Dateien (raw gelesen, Referenz Genomsequenz Annotation Liste) korrekt formatiert sind. Im zweiten Schritt VORVERARBEITUNG (Abbildung 5A - orange Box), minderwertige Sequenzierungsdaten sind zunächst getrimmt mit Cutadapt28 abhängig von Qualitätskennzahlen und liest mit weniger als 100 Basen Länge werden verworfen. In einem weiteren Schritt lesen Ausrichtung (Abbildung 5A - grüner Kasten) sind die restlichen liest Blastn29 , die Genomsequenz ermöglicht bis zu 5 % von Konflikten ausgerichtet. Eine SAM-Datei wird erstellt und liest nur mit Qualitätsfaktor größer als 30 (Q > 30) werden verarbeitet mit SAMtools30 und in einer BAM-Datei umgewandelt. Nach dem ausrichten Interaktom-Seq führt die Domänen-Erkennung (Abbildung 5A - Blue-Box), Aufrufen von Bedtools31 Filtern liest Überlappung mindestens 80 % ihrer Länge in Abschriften; Abdeckung, max. Tiefe und Fokus-Werte werden dann für jeden ORF Teil abgedeckt durch die Zuordnung lautet berechnet. Die Abdeckung ist die Gesamtzahl der Lesevorgänge zu einem Gen zugeordnet; die Tiefe beträgt die maximale Anzahl der Lesevorgänge für eine spezifische genetische Teil; der Fokus ist ein Index, gewonnen aus dem Verhältnis zwischen max Tiefe und Abdeckung, und es kann im Bereich zwischen 0 und 1. Wenn der Fokus höher als 0,8 ist und die Abdeckung höher als die durchschnittliche Abdeckung für alle Regionen der Zuordnung in der BAM-Datei beobachtet ist, wird der CDS-Teil als eine vermeintliche Domäne/Epitop eingestuft. Der letzte Schritt der Interaktom-Seq-Pipeline ist die Ausgabe (Abbildung 5A - violetten Kasten), eine Liste von vermeintlichen Domains im Tabellenformat getrennt erzeugt wird. Die Interaktom-Seq-Pipeline wurde in ein Web-Tool, damit Benutzer ohne Programmierkenntnisse oder Bioinformatik Interaktom-Seq-Analyse über die grafische Benutzeroberfläche durchführen und ihre Ergebnisse in einem einfachen und benutzerfreundlichen Format aufgenommen. Wie in Abbildung 5 bgezeigt, werden die ausgegebenen Ergebnisse einer Analyse mit JBrowse32 , Visualisierung und Exploration ermöglichen angezeigt. Interaktom-Seq erzeugt Spuren im Genom Browser entsprechend vermeintliche Domänen erkannt und bietet auch klassische Venn-Diagramme um Kreuzungen zwischen gemeinsamen vermeintliche Domänen bereichert z.B. in verschiedenen Auswahlen Experimente zeigen.

Abbildung 1: Schematische Übersicht über die wichtigsten Schritte für den Bau der ORF-Filterung Bibliothek
(A) DNA aus anderen Quelle ist beschallt und fragmentiert in zufällige Fragmente von 150-750 bp Länge. Fragmente sind erholte sich von Gel und als Stumpf in der pFILTER-Vektor kloniert. (B) Filterung Schritt mit β-Lactamase als klappbare Reporter. Vektor, enthält nicht ORF Fragmente sind negativ ausgewählt auf Ampicillin beim ORF geklonten Fragmente können Kolonien wachsen; (C) Anwendung von einer zunehmenden Selektionsdruck (Ampicillin Konzentration in solides Wachstumsmedien von 0 bis > 100 μg/mL) ermöglicht die Auswahl der besser gefalteten Fragmente. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2: Schematische Übersicht über die wichtigsten Schritte für den Bau der Phagen-Bibliothek
(A) ORF-gefilterte Fragmente werden aus dem gefilterten Vektor verwenden bestimmte Restriktionsenzyme ausgeschnitten. Nach Wiederherstellung und Reinigung sind Fragmente in Phagemid Vektor kloniert und verwandelt; (B) Phagemid bakterielle Bibliothek mit Helfer Phagen infiziert ist und nach Übernachtung Wachstum Phagen sind PEG ausgefällt und gesammelt. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3: ORF Bibliotheken Sequenzierung
Sequenzierung erfolgt auf beiden ursprünglichen ORF ausgewählten Bibliothek sowie die Phagen-Display-Bibliothek; (1) auf beiden Fällen Kolonien gewachsen zurückgewonnen und DNA extrahiert; (2) DNA-Fragmente werden durch Amplifikation mit spezifischen Primer verbunden mit Adaptern für die Sequenzierung wiederhergestellt; 3-4) Fragmente sind erholt und tiefen sequenziert mit NGS; (5) Daten werden mithilfe der Interaktom-Seq-Pipeline analysiert. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 4: Schematische Übersicht der Bibliothek Auswahl mit Patienten Antikörper
Phagen-Bibliothek ist für die Auswahl gegen Antikörper von Patienten Sera verwendet. Antikörper sind auf magnetischen Kügelchen immobilisiert, Phagen-Bibliothek-Erfassung/Selektion erfolgt, drei Zyklen der Waschgänge durchgeführt und danach ausgewählte Phagen wiedergewonnen und erneut infizieren zur E. Coli. Wieder infiziert E. Coli Zellen sind in selektiven Druck (Ampicillin 100 μg/mL) beschichtet. ORF-Fragmente werden durch Amplifikation wiederhergestellt und Amplifikate Pools sind dann von NGS sequenziert. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 5: Schematische Übersicht über Bibliotheksanalyse
(A) die Darstellung des Daten-Analyse-Workflows, raw FASTQ-Dateien ab, bis die endgültige kommentierte Domänen-Listen; (B) schematische Darstellung der ein- und Ausgänge das Interaktom-Seq-Web-Tool. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
Diskussion
Die Schaffung einer qualitativ hochwertige unterschiedlichsten ORFs gefilterte Bibliothek ist der erste wichtige Schritt in dem gesamten Verfahren, da sie die nachfolgenden Schritte der Pipeline beeinflussen.
Ein wichtiges Merkmal der Vorteil unserer Methode ist, dass jede Quelle (intronless) DNA (cDNA, genomische DNA, PCR abgeleitet oder synthetische DNA) geeignet für Bibliotheksbau. Der erste Parameter, der berücksichtigt werden sollte ist, dass die Länge der DNA-Fragmente in der pFILTER-Vektor kloniert sollte eine Darstellung der gesamten Sammlung der Domains ein Genom oder einer Transkriptom, die so genannte "Domainome". Wir haben gezeigt, dass Protein Domains können erfolgreich geklont werden, ausgewählt und schließlich identifiziert ausgehend von DNA-Fragmente mit einer Länge Verteilung überspannt von 150 bis 750 bp33,34, und dies ist im Einklang mit dem, was in gemeldet wird die Literatur zeigt, dass die meisten Protein-Domains 100 aa lang sind (mit einem Bereich von 50 bis 200 aa)15.
DNA Ausgangsmaterial muss in den Größenbereich von Wahl und später in den Filter (pFILTER)12 Vektor geklonten fragmentiert werden. Während dieser Schritte könnten potenzielle Verzerrung Maximierung der Effizienz aller das Klonen Reaktionen in das Protokoll, in bestimmten Fragment Ende-Reparatur und Phosphorylierung enthalten Schritte vermieden werden. Die Vektor-Vorbereitung ist anspruchsvoll und erfolgen unter optimalen Bedingungen als auch Plasmid Abbau und/oder Verschmutzung durch unverdaute Vektor zu vermeiden.
Nachdem die Bibliothek erstellt wurde, sollte es "gefiltert werden," um nur ORFs gefalteten Fragmente erhalten. Ein wichtiger Parameter zu modulieren, dieser Schritt ist die selektive Druck angewendet, die nach der Strenge der Filterung auf Wunsch geändert werden kann. Auswahl erfolgt mit Ampicillin: je höhere der Konzentration verwendet, desto geringere die Anzahl der transformierten Bakterien-Kolonien in der Lage zu überleben. Dies spiegelt die Fähigkeit der Filtermethode für gut-im Vergleich zu Armen-Ordner ORFs34wählen. Diese Reduktion in der Anzahl der Klone wird durch die Erhöhung der Eigenschaften des ausgewählten Fragmente Falten ausgeglichen. In der Regel sollte die Ampicillin-Konzentration ausreichen um auf etwa 1/20 die Anzahl der Bakterienkolonien im Hinblick auf diejenigen reduzieren, die wachsende Bibliothek auf Chloramphenicol nur erzielt werden konnten.
Bibliothek-Validierung erfolgt in der Regel durch PCR-Amplifikation von zufällig ausgewählten Kolonien und deren Sequenzierung. PCR-Amplifikation von einigen Kolonien wird vorgeschlagen, um eine schnelle Einschätzung der Qualität der Bibliothek zu haben: die Länge der Einsätze sollte im erwarteten Bereich von 150-750 bp und verschiedenen Kolonien sollte vorhanden Einsätze mit verschiedenen Größe anzeigt gut Bibliothek-Vorbereitung in Bezug auf die Variabilität. Diese herkömmliche Strategie des Screenings, als die einzige Methode für die Bibliothek Validierung aufgetragen ist nicht vollständig und ist zeitaufwendig, erlaubt die Analyse nur eine begrenzte Anzahl von Kolonien und mit hoher Wahrscheinlichkeit die meisten wichtigen Klone zu verpassen. Unser Ansatz basiert auf Tiefe Sequenzierung der Bibliothek, dies bietet umfassende Informationen über die Bibliothek Vielfalt und Fülle und genaue Kartierung der jedes der ausgewählten Fragmente.
Die Umsetzung der NGS-Technologie mit der Filterung Ansatz erhöht die Tiefe der Analyse um mehrere Größenordnungen. Wir haben vor kurzem das Protokoll für die Sequenzierung der ORF-Bibliotheken mithilfe der Illumina-Plattform optimiert und entwickelt eine bestimmte Web-Tool für die Datenanalyse, die die Analyse dieser Art von Daten für jeden Nutzer ohne irgendwelche Programmierkenntnisse Bioinformatik macht.
Die Bibliothek "per se" ist ein "universelles Instrument" und kann in unterschiedlichen Kontexten für Protein-Expression und/oder Auswahl genutzt werden. Unsere methodische Ansatz basiert auf den produzierten ORFeome in einem Phagen-Display-Kontext übertragen. PROTEINFRAGMENTE werden auf der Oberfläche von Phagen ausgedrückt und wurde für nachfolgende Auswahl geeignet.
Dies erfolgt durch Rettung der gefilterten ORFs aus der pFILTER-Bibliothek durch Verdauung mit speziellen Restriktionsenzymen und Klonen sie erneut in einen kompatiblen Phagemid Vektor so dass ihre Verschmelzung mit der Phagen Protein g3p.
Nachdem die Phagemid-ORF-Bibliothek erstellt wurde, kann es für die Auswahl gegen verschiedene Ziele, z. B. eine vermeintliche Bindung Protein10 oder gereinigte Antikörper35,36 wie hier beschrieben verwendet werden. Da Phagen Partikel angezeigt werden, auf ihrer Oberfläche, das gefilterte ORFs, daraus resultiert eine wesentlich effektiver Auswahlverfahren aufgrund der Abwesenheit nicht anzeigen-Klone in der Regel überholen sie.
Nach der Auswahl der Phage Display ORF Bibliothek können mit der gleichen Rohrleitung die Ausgabe Klone sequenziert und analysiert werden. NGS bieten eine komplette und statistisch signifikante Ranking der am meisten häufig ausgewählt ORFs und dies erlaubt die Identifikation von Proteinen, die meist Interaktion mit dem Köder verwendet. Angesichts der Präsenz von vielen verschiedenen Versionen der jeweiligen Domäne unterscheiden sich durch einige Aminosäuren, identifiziert die Überschneidungen zwischen verschiedenen sequenzierten Klonen auch die minimale Fragment/Domäne zeigt Bindungseigenschaften. Schließlich durch die Kopplung von Genotyp und Phänotyp Informationen in die Phagen-Bibliothek, kann sobald die Domains Wahl identifiziert wurden, die DNA-Sequenz leicht aus der Bibliothek für weitere Studien, in-Vitro und in Vivo gerettet werden Validierung und Charakterisierung.
Offenlegungen
Die Autoren haben nichts preisgeben.
Danksagungen
Diese Arbeit wurde unterstützt durch einen Zuschuss aus dem italienischen Ministerium für Bildung und Universität (2010P3S8BR_002, CP).
Materialien
| Name | Company | Catalog Number | Comments |
| Sonopuls ultrasonic homogenizer | Bandelin | HD2070 | or equivalent |
| GeneRuler 100 bp Plus DNA Ladder | Thermo Scientific | SM0321 | or equivalent |
| GeneRuler 1 kb DNA Ladder | Thermo Fisher Scientific | SM0311 | or equivalent |
| Molecular Biology Agarose | BioRad | 161-3102 | or equivalent |
| Green Gel Plus | Fisher Molecular Biology | FS-GEL01 | or equivalent |
| 6x DNA Loading Dye | Thermo Fisher Scientific | R0611 | or equivalent |
| QIAquick Gel Extraction Kit | Qiagen | 28704 | or equivalent |
| Quick Blunting Kit | New England Biolabs | E1201S | |
| NanoDrop 2000 UV-Vis Spectrophotometer | Thermo Fisher Scientific | ND-2000 | |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | 4368813 | |
| Streptavidin Magnetic Beads | New England Biolabs | S1420S | or equivalent |
| QIAquick PCR purification Kit | Qiagen | 28104 | or equivalent |
| EcoRV | New England Biolabs | R0195L | |
| Antarctic Phosphatase | New England Biolabs | M0289S | |
| T4 DNA Ligase | New England Biolabs | M0202T | |
| Sodium Acetate 3M pH5.2 | general lab supplier | ||
| Ethanol for molecular biology | Sigma-Aldrich | E7023 | or equivalent |
| DH5aF' bacteria cells | Thermo Fisher Scientific | ||
| 0,2 ml tubes | general lab supplier | ||
| 1,5 ml tubes | general lab supplier | ||
| 0,1 cm electroporation cuvettes | Biosigma | 4905020 | |
| Electroporator 2510 | Eppendorf | ||
| 2x YT medium | Sigma-Aldrich | Y1003 | |
| Ampicillin sodium salt | Sigma-Aldrich | A9518 | |
| Chloramphenicol | Sigma-Aldrich | C0378 | |
| DreamTaq DNA Polymerase | Thermo Fisher Scientific | EP0702 | |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | N0447S | |
| 96-well thermal cycler (with heated lid) | general lab supplier | ||
| 150 mm plates | general lab supplier | ||
| 100 mm plates | general lab supplier | ||
| Glycerol | Sigma-Aldrich | G5516 | |
| BssHII | New England Biolabs | R0199L | |
| NheI | New England Biolabs | R0131L | |
| QIAprep Spin Miniprep Kit | Qiagen | 27104 | or equivalent |
| M13KO7 Helper Phage | GE Healthcare Life Sciences | 27-1524-01 | |
| Kanamycin sulfate from Streptomyces kanamyceticus | Sigma-Aldrich | K1377 | |
| Polyethylene glycol (PEG) | Sigma-Aldrich | P5413 | |
| Sodium Cloride (NaCl) | Sigma-Aldrich | S3014 | |
| PBS | general lab supplier | ||
| Dynabeads Protein G for Immunoprecipitation | Thermo Fisher Scientific | 10003D | or equivalent |
| MagnaRack Magnetic Separation Rack | Thermo Fisher Scientific | CS15000 | or equivalent |
| Tween 20 | Sigma-Aldrich | P1379 | |
| Nonfat dried milk powder | EuroClone | EMR180500 | |
| KAPA HiFi HotStart ReadyMix | Kapa Biosystems, Fisher Scientific | 7958935001 | |
| AMPure XP beads | Agencourt, Beckman Coulter | A63881 | |
| Nextera XT dual Index Primers | Illumina | FC-131-2001 or FC-131-2002 or FC-131-2003 or FC-131-2004 | |
| MiSeq or Hiseq2500 | Illumina | ||
| Spectrophotomer | Nanodrop | ||
| Agilent Bioanalyzer or TapeStation | Agilent | ||
| Forward PCR primer | general lab supplier | 5’ TACCTATTGCCTACGGCA GCCGCTGGATTGTTATTACTC 3’ | |
| Reverse PCR primer | general lab supplier | 5’ TGGTGATGGTGAGTACTA TCCAGGCCCAGCAGTGGGTTTG 3’ | |
| Forward primer for NGS | general lab supplier | 5’ TCGTCGGCAGCGTCAGA TGTGTATAAGAGACAGGCA GCAAGCGGCGCGCATGC 3’; | |
| Reverse primer for NGS | general lab supplier | 5’ GTCTCGTGGGCTCGGAGA TGTGTATAAGAGACAGGGG ATTGGTTTGCCGCTAGC 3’; |
Referenzen
- Loman, N. J., Pallen, M. J. Twenty years of bacterial genome sequencing. Nat Rev Microbiol. 13 (12), 787-794 (2015).
- Jones, C. E., Brown, A. L., Baumann, U. Estimating the annotation error rate of curated GO database sequence annotations. BMC Bioinformatics. 8 (1), 170(2007).
- Andorf, C., Dobbs, D., Honavar, V. Exploring inconsistencies in genome-wide protein function annotations: a machine learning approach. BMC Bioinformatics. 8 (1), 284(2007).
- Wong, W. -C., Maurer-Stroh, S., Eisenhaber, F. More Than 1,001 Problems with Protein Domain Databases: Transmembrane Regions, Signal Peptides and the Issue of Sequence Homology. PLoS Comput Biol. 6 (7), e1000867(2010).
- Bioinformatics, B., et al. Identification and correction of abnormal, incomplete and mispredicted proteins in public databases. BMC Bioinformatics. 9 (9), (2008).
- Phizicky, E., Bastiaens, P. I. H., Zhu, H., Snyder, M., Fields, S. Protein analysis on a proteomic scale. Nature. 422 (6928), 208-215 (2003).
- DiDonato, M., Deacon, A. M., Klock, H. E., McMullan, D., Lesley, S. A. A scaleable and integrated crystallization pipeline applied to mining the Thermotoga maritima proteome. J Struct Funct Genomics. 5 (1-2), 133-146 (2004).
- Nordlund, P., et al. Protein production and purification. Nat Methods. 5 (2), 135-146 (2008).
- Zacchi, P., Sblattero, D., Florian, F., Marzari, R., Bradbury, A. R. M. Selecting open reading frames from DNA. Genome Res. 13 (5), 980-990 (2003).
- Di Niro, R., et al. Rapid interactome profiling by massive sequencing. Nucleic Acids Res. 38 (9), e110(2010).
- Gourlay, L. J., et al. Selecting soluble/foldable protein domains through single-gene or genomic ORF filtering: Structure of the head domain of Burkholderia pseudomallei antigen BPSL2063. Acta Crystallogr Sect D Biol Crystallogr. 71 (Pt 11), 2227-2235 (2015).
- D'Angelo, S., et al. Filtering "genic" open reading frames from genomic DNA samples for advanced annotation. BMC Genomics. 12 (Suppl 1), S5(2011).
- D'Angelo, S., et al. Profiling celiac disease antibody repertoire. Clin Immunol. 148 (1), 99-109 (2013).
- Robinson, M. D., McCarthy, D. J., Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26 (1), 139-140 (2009).
- Heger, A., Holm, L. Exhaustive enumeration of protein domain families. J Mol Biol. 328 (3), 749-767 (2003).
- Zacchi, P., Sblattero, D., Florian, F., Marzari, R., Bradbury, A. R. M. Selecting open reading frames from DNA. Genome Res. 13 (5), 980-990 (2003).
- Faix, P. H., Burg, M. A., Gonzales, M., Ravey, E. P., Baird, A., Larocca, D. Phage display of cDNA libraries: Enrichment of cDNA expression using open reading frame selection. Biotechniques. 36 (6), 1018-1029 (2004).
- Patrucco, L., et al. Identification of novel proteins binding the AU-rich element of α-prothymosin mRNA through the selection of open reading frames (RIDome). RNA Biol. 12 (12), 1289-1300 (2015).
- Collins, M. O., Choudhary, J. S. Mapping multiprotein complexes by affinity purification and mass spectrometry. Curr Opin Biotechnol. 19 (4), 324-330 (2008).
- Suter, B., Kittanakom, S., Stagljar, I. Two-hybrid technologies in proteomics research. Curr Opin Biotechnol. 19 (4), 316-323 (2008).
- Nakai, Y., Nomura, Y., Sato, T., Shiratsuchi, A., Nakanishi, Y. Isolation of a Drosophila gene coding for a protein containing a novel phosphatidylserine-binding motif. J Biochem. 137 (5), 593-599 (2005).
- Deng, S. J., et al. Selection of antibody single-chain variable fragments with improved carbohydrate binding by phage display. J Biol Chem. 269 (13), 9533-9538 (1994).
- Danner, S., Belasco, J. G. T7 phage display: A novel genetic selection system for cloning RNA-binding proteins from cDNA libraries. Proc Natl Acad Sci. 98 (23), 12954-12959 (2001).
- Gargir, A., Ofek, I., Meron-Sudai, S., Tanamy, M. G., Kabouridis, P. S., Nissim, A. Single chain antibodies specific for fatty acids derived from a semi-synthetic phage display library. Biochim Biophys Acta - Gen Subj. 1569 (1-3), 167-173 (2002).
- Patrucco, L., et al. Identification of novel proteins binding the AU-rich element of α-prothymosin mRNA through the selection of open reading frames (RIDome). RNA Biol. 12 (12), 1289-1300 (2015).
- Ausubel, F. M., et al. Current Protocols in Molecular Biology. Mol Biol. 1 (2), 146(2003).
- Sblattero, D., Bradbury, A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat Biotechnol. 18, 75-80 (2000).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 17 (1), 10(2011).
- Camacho, C., et al. BLAST+: architecture and applications. BMC Bioinformatics. 10 (1), 421(2009).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Quinlan, A. R. BEDTools: The Swiss-Army tool for genome feature analysis. Curr Protoc Bioinforma. , (2014).
- Skinner, M. E., Uzilov, A. V., Stein, L. D., Mungall, C. J., Holmes, I. H. JBrowse: A next-generation genome browser. Genome Res. 19 (9), 1630-1638 (2009).
- Gourlay, L. J., et al. Selecting soluble/foldable protein domains through single-gene or genomic ORF filtering: Structure of the head domain of Burkholderia pseudomallei antigen BPSL2063. Acta Crystallogr Sect D Biol Crystallogr. 71, 2227-2235 (2015).
- D'Angelo, S., et al. Filtering "genic" open reading frames from genomic DNA samples for advanced annotation. BMC Genomics. 12 (Suppl 1), S5(2011).
- Di Niro, R., et al. Characterizing monoclonal antibody epitopes by filtered gene fragment phage display. Biochem J. 388 (Pt 3), 889-894 (2005).
- D'Angelo, S., et al. Profiling celiac disease antibody repertoire. Clin Immunol. 148 (1), 99-109 (2013).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten