
Detecção de Microrganismos Ambientais com Reação em Cadeia da Polimerase e Eletroforese em Gel
Visão Geral
Fonte: Laboratórios do Dr. Ian Pepper e Dr. Charles Gerba - Universidade do Arizona
Autor de Demonstração: Bradley Schmitz
A reação em cadeia de polimerase (PCR) é uma técnica usada para detectar microrganismos presentes em ambientes de solo, água e atmosfera. Ao amplificar seções específicas de DNA, o PCR pode facilitar a detecção e identificação de microrganismos-alvo até a espécie, cepa e nível de sorovar/pathovar. A técnica também pode ser utilizada para caracterizar comunidades inteiras de microrganismos em amostras.
A criação de microrganismos em laboratório utilizando mídia especializada de crescimento é uma técnica há muito estabelecida e permanece em uso para a detecção de microrganismos em amostras ambientais. Muitos micróbios no ambiente natural, embora vivos, mantêm baixos níveis de atividade metabólica e/ou tempos de duplicação e são, portanto, referidos como organismos viáveis, mas não culturais (VBNC). O uso de técnicas baseadas na cultura por si só não pode detectar esses micróbios e, portanto, não fornece uma avaliação minuciosa das populações microbianas em amostras. O uso de PCR permite a detecção de micróbios culturais, organismos VBNC e aqueles que não estão mais vivos ou ativos, pois a amplificação de sequências genéticas geralmente não requer o pré-enriquecimento de microrganismos presentes em amostras ambientais. No entanto, o PCR não pode diferenciar os estados acima mencionados de viabilidade e atividade. Quando combinada com uma ou mais técnicas baseadas em cultura, a viabilidade de certos subconjuntos de microrganismos ainda pode ser determinada.
Princípios
A premissa básica do PCR é usar ciclos repetidos de mudanças de temperatura sequenciais para alcançar a amplificação exponencial do DNA. A síntese de DNA é realizada por enzimas de polimerase de DNA que são obtidas de bactérias que vivem em fontes termais, como thermus aquaticus (Taq). Estas polimerases são estáveis em calor, permitindo-lhes suportar as altas temperaturas utilizadas durante a PCR.
A sequência de destino, conhecida como amplicon, é amplificada a partir do modelo de DNA usando dois pequenos trechos de nucleotídeos conhecidos como "primers". Devido à alta especificidade da ligação de ácido nucleico complementar, os primers permitem a amplificação direcionada de sequências de interesse muito específicas. Ao projetar primers que apenas amplificarão uma sequência única (ou uma combinação única de sequências) de um organismo de interesse, o PCR pode ser usado para detectar diferencialmente para a presença do DNA desse organismo entre todos os materiais genéticos presentes em uma amostra ambiental complexa.
Para executar o PCR, uma máquina conhecida como termociclador é usada para pedalar automaticamente através das diferentes temperaturas necessárias para a reação. Cada ciclo é dividido em três fases. O primeiro, conhecido como "desnaturação", é geralmente definido acima de 92 °C e dura cerca de 30 s. A desnaturação é usada para quebrar moléculas de DNA em fios únicos, para permitir que a reação de amplificação prossiga.
A segunda fase, "ressarida", é definida 2-3 °C abaixo da temperatura de fusão dos dois primers, geralmente entre 50-65 °C, e também dura cerca de 30 s. A temperatura de fusão é a temperatura em que 50% do DNA de duplaridade se separou em fios únicos, e assim o passo de ressaramento permite que os primers se liguem aos seus locais alvo no modelo de DNA.
A terceira fase de um ciclo PCR é "alongamento" ou "extensão", quando a polimerase de DNA se liga ao duplex de modelo de primer e catalisa a síntese do produto. Fixada em 72 °C para a polimerase Taq, a duração desta fase depende do comprimento do amplicon, geralmente 30 s / 500 bp. Após cada ciclo, o DNA amplificado é mais uma vez desnaturado e serve como um novo modelo, levando a um aumento exponencial na quantidade de produtos PCR.
Uma vez que a reação esteja completa, os produtos PCR podem ser resolvidos pelo tamanho de um "gel" geralmente feito da agarose do polímero, um processo conhecido como eletroforese. Um campo elétrico é aplicado através do gel, e as cargas negativas na espinha dorsal das moléculas de DNA fazem com que elas migrem para o lado positivo do campo. De um modo geral, moléculas de DNA linear que são maiores levarão mais tempo para viajar através da matriz de gel.
Procedimento
1. Coleta de Amostras
- Colete o solo usando um auger ou pá até uma profundidade determinada. Se coletar o solo da rizosfera (a região estreita do solo ao redor e influenciada pelas raízes das plantas e seus micróbios associados), basta coletar diretamente do entorno das raízes da planta, atingindo o solo em um barril de coleta.
- Colete a amostra de água mergulhando uma garrafa de plástico estéril na água enquanto segura a ponta do bastão de mergulho.
2. Extração e Preparação de Ácidos Nucleicos
- Coletar organismos e vírus da amostra, e extrair DNA e RNA deles. Para obter detalhes, consulte o vídeo da JoVE Science Education sobre extração de ácido nucleico comunitário.
- Armazene o DNA extraído em tubos de microfudão rotulados. Se o DNA precisar ser armazenado durante a noite ou por períodos mais longos de tempo, congele-o a -20 °C e descongele os tubos à temperatura ambiente quando estiver pronto para uso.
- Se o material genético a ser avaliado for RNA (seja o genoma dos vírus RNA, ou o RNA transcrito de organismos celulares), realize transcrição reversa (RT) na amostra para criar DNA complementar (cDNA) antes de prosseguir para pcr. Para obter detalhes, consulte o vídeo da JoVE Science Education no RT-PCR.
3. Reação em cadeia de polimerase
- Coloque a enzima PCR (por exemplo,polímerase de Taq) no gelo e descongele os outros reagentes (tampão PCR, dNTPs, primers) dentro de um capô "limpo" designado à temperatura ambiente. A enzima é armazenada a -20 °C, mas nunca congela. É sensível à temperatura, e por isso deve ser mantido frio e sua exposição à temperatura ambiente minimizada
- Calcule o volume de cada reagente necessário para fazer uma "mistura mestre" de todos os reagentes que são constantes entre todas as reações(Tabela 1). Certifique-se de contabilizar controles positivos (por exemplo,um modelo conhecido por conter a região alvo) e negativos(por exemplo,sem modelo) nos cálculos. Adicione um adicional de 10% ao volume final para explicar o erro de pipetação. Os volumes de primer dependem de ensaios para organismos específicos; referem-se à literatura publicada para os valores apropriados.
- Usando um tubo de microfuça de baixa ligação, que minimiza a adesão de moléculas de reagente às paredes plásticas, adicione os volumes calculados de cada reagente para montar a mistura mestre. Vórtice suavemente e centrífuga cada reagente antes de adicionar. Uma vez que a mistura mestre é preparada, vórtice para misturar e coletar por centrifugação
- Prepare tiras PCR de 8 tubos. Designe um tubo para cada amostra, incluindo controles positivos e negativos
- Dispense o volume apropriado da mistura PCR em cada tubo da tira
- Adicione o volume apropriado de modelo de DNA das amostras, bem como 5 μL do modelo positivo e 5 μL de água de grau molecular como controle negativo, nos respectivos tubos PCR.
- Coloque a tampa com segurança na tira de 8 tubos e centrífuga por alguns segundos usando uma minicentrifuge
- Coloque a tira de 8 tubos em um termociclador
- Defina o programa PCR apropriado para ser executado no termociclador. Um programa típico consiste no seguinte
- Denaturação a 94 °C por 3 min.
- 30-40 ciclos de amplificação: desnaturação a 94 °C para 30 s, ressarem a temperatura específica do primer (geralmente entre 50-60 °C) para 30 s, e extensão a 72 °C para 30 s / 500 bp.
- Prorrogação final a 72 °C para 7-10 min.
4. Preparação do Gel Agarose
- Com base no volume de gel desejado e concentração de gel(Tabela 2),pesa a quantidade apropriada de pó de agarose em um frasco de Erlenmeyer de 125 mL.
- Adicione o volume apropriado de tampão de gel no frasco e gire o frasco à mão.
- Aqueça a mistura tampão-agarose em um forno de micro-ondas em alta potência por 1 min.
- Remova o frasco do micro-ondas e gire à mão para garantir que toda a agarose tenha dissolvido. Se a agarose não tiver se dissolvido completamente, repita a microwaving em incrementos de 30 s.
- Depois de fixar firmemente a tampa no frasco, esfrie-a a 50 °C girando sob água fria.
- Adicione 1 μL de brometo de etídio (EtBr) à mistura de agarose usando uma micropippette designada para EtBr. EtBr é um corante que une ácidos nucleicos de dupla vertente e laranja fluoresces quando iluminado com luz UV. Observe que o EtBr é potencialmente cancerígeno, por isso devem ser usados equipamentos de proteção individual (óculos, jaleco, luvas resistentes ao EtBr).
- Despeje o gel derretido em uma bandeja de fundição de gel de eletroforese. Certifique-se de que nenhuma bolha está presa dentro da agarose. Coloque um pente no gel e aperte com segurança. Espere aproximadamente 20-30 min para o gel se solidificar.
- Uma vez que o gel tenha solidificado, remova o pente cuidadosamente sem causar lágrimas no gel. O pente cria poços no gel para carregar as amostras.
5. Eletroforese de gel
- Coloque o gel de agarose solidificado na câmara de eletroforese.
- Adicione o buffer de corrida apropriado na câmara até que o gel mal esteja submerso.
- Em um pedaço de Parafilm, manchas de pipeta de concentrado de corante de carga. Inclua também uma escada de DNA de uma gama adequada para os tamanhos esperados dos produtos PCR. Alternativamente, use um conjunto fresco de tubos de microfuge, um para cada amostra.
- Uma vez que o PCR esteja concluído, recupere essa tira de 8 tubos do termociclador e centrífugas brevemente para coletar quaisquer condensados. Adicione um volume apropriado de produto de DNA ao corante de carregamento e pipeta para cima e para baixo para misturar. Por exemplo, 2 μL de corante de carregamento de 10x é misturado com 8 μL de amostra para dar um volume final de 10 μL, com o corante em 1x.
- Pipeta cada mistura de amostra de corante nos poços designados no gel de agarose, tomando cuidado para não perfurar os poços.
- Ligue os eletrodos à câmara de eletroforese. O DNA é carregado negativamente, por isso "corre" em direção ao eletrodo positivo. Portanto, conecte o eletrodo positivo ao lado oposto da câmara, onde os poços foram carregados. Defina a fonte de alimentação para uma tensão apropriada para o sistema tampão e o tamanho da câmara de gel e ajuste-a para funcionar. Pequenas bolhas devem ser visíveis movendo-se para os lados da câmara se a eletroforese estiver procedendo corretamente.
- Uma vez que a frente de corante tenha avançado o suficiente para baixo do gel, desligue a fonte de alimentação. Transporte cuidadosamente o gel para o transilluminador ou imagem visual e ligue a luz UV para visualizar as bandas de DNA no gel.
- Analise o tamanho da banda e as posições do gel. Compare as posições de banda das amostras com o controle positivo para determinar se o DNA do organismo de interesse está presente na amostra (Figura 1).
| Componente | Volume por tubo(μL) | Volume para 5 tubos(μL) | Concentração Final |
| Tampão 10x Ex Taq | 5.0 | 25 | 1x |
| DNTPs de 2,5 mM | 4.0 | 20 | 0,2 mM |
| Primer forward* | 2.0 | 10 | 400 nM |
| Primer reverso* | 2.0 | 10 | 400 nM |
| Molecular H2O | 31.75 | 158.75 | - |
| Ex Taq | 0.25 | 1.25 | 2.5 U |
| Mistura PCR | 45 | 225 |
Mesa 1. Volumes de reagentes para mix mestre pcr. *Os volumes do primer variam dependendo do ensaio do organismo. Ajuste o volume de água de grau molecular para fazer o volume final 45 μL. Os volumes de outros componentes não devem variar.
| % recomendado de Agarose | Resolução ideal para fragmentos lineares de DNA (pares de base) |
| 0.5 | 1,000-30,000 |
| 0.7 | 800-12,000 |
| 1.0 | 500-10,000 |
| 1.2 | 400-7,000 |
| 1.5 | 200-3,000 |
| 2.0 | 50-2,00 |
Mesa 2. O tamanho do fragmento de DNA varia idealmente resolvido por diferentes porcentagens de gel de agarose.
Resultados
Na Figura 1,a escada de DNA (faixa 1) fornece uma referência para o tamanho e concentração aproximada para bandas dos produtos PCR. O controle negativo (pista 2) não contém nenhum material genético, enquanto o controle positivo (pista 3) é amplificado a partir de modelos conhecidos por conter o DNA alvo para indicar tamanho e localização de bandas alvo. As amostras 4, 6, 8 e 9 apresentam padrão de banda semelhante ao controle positivo, indicando, portanto, que essas amostras contêm o material genético alvo. Pode-se inferir que o organismo está presente nos ambientes a partir dos quais essas amostras foram obtidas.
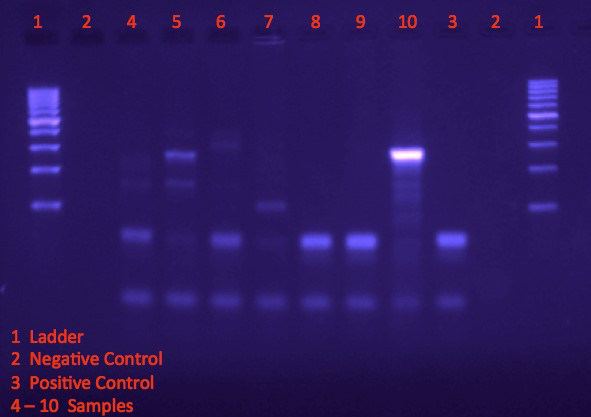
Figura 1. Visualizando bandas em gel de agarose após eletroforese.
Aplicação e Resumo
O PCR pode ser empregado para determinar rapidamente a presença ou ausência de patógenos no ambiente. Por exemplo, primers específicos da ameba comedora de cérebro, Naegleria fowleri, amplificarão o DNA e produzirão bandas fortes em um gel se o organismo estiver presente em uma amostra. Se um único organismo não é o principal interesse, mas sim genes associados à produção de toxinas de uma variedade de organismos, o PCR também pode ser usado para determinar a presença ou ausência desses materiais genéticos específicos.
O PCR também pode ser usado como procedimento de confirmação ao analisar micróbios ambientais em laboratório. Se um método de cultura não pode diferenciar entre certos organismos que estão presentes em uma amostra ambiental, então o PCR talvez tenha sido usado para distinguir especificamente entre os micróbios candidatos.
O PCR convencional pode ser modificado de várias maneiras para fins experimentais específicos. O PCR pode ser usado para analisar modelos de RNA de fio único, acoplados a uma etapa de transcrição reversa (RT-PCR). Além da determinação da presença versus ausência, a PCR quantitativa (qPCR) pode medir a concentração para DNA específico de interesse.
Pular para...
Vídeos desta coleção:

Now Playing
Detecção de Microrganismos Ambientais com Reação em Cadeia da Polimerase e Eletroforese em Gel
Environmental Microbiology
44.6K Visualizações

Determinação do teor de umidade no solo
Environmental Microbiology
359.7K Visualizações

Técnica Asséptica em Ciências Ambientais
Environmental Microbiology
126.5K Visualizações

Coloração de Gram de bactérias de fontes ambientais
Environmental Microbiology
100.5K Visualizações

Visualização de Microrganismos do Solo por Meio do Ensaio de Lâmina de Contato e Microscopia
Environmental Microbiology
42.4K Visualizações

Fungos Filamentosos
Environmental Microbiology
57.4K Visualizações

Extração de DNA de comunidades de colônias bacterianas
Environmental Microbiology
28.9K Visualizações

Análise de RNA de amostras ambientais usando RT-PCR
Environmental Microbiology
40.4K Visualizações

Quantificando Microorganismos e Vírus Ambientais Usando qPCR
Environmental Microbiology
47.9K Visualizações

Análise da Qualidade da Água por Organismos Indicadores
Environmental Microbiology
29.6K Visualizações

Isolamento de bactérias fecais de amostras de água por filtração
Environmental Microbiology
39.4K Visualizações

Detecção de Bacteriófagos em Amostras Ambientais
Environmental Microbiology
40.8K Visualizações

Cultivo e Enumeração de Bactérias de Amostras de Solo
Environmental Microbiology
184.8K Visualizações

Análise da Curva de Crescimento Bacteriano e suas Aplicações Ambientais
Environmental Microbiology
296.2K Visualizações

Contagem de Algas em Métodos de Cultura
Environmental Microbiology
13.8K Visualizações
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados