
Method Article
レンチ ウイルス媒介トランスジェニック マウスの生産: シンプルで高効率な創設者の直接の研究法
要約
ここでは、受精卵母細胞の囲卵腔のレンチウイルスベクターの単純な注射によって transgene の統合および創設者トランスジェニック マウスに高効率の生産を促進するためにプロトコルを提案する.
要約
ほぼ 40 年間は、前核 DNA 注入はトランスジェニック マウスに遺伝子のランダムな統合を生成する標準的な方法を表します。このようなルーチンの手順が世界中で広く利用されて、transgene の統合の創設者動物の低収量の結果の悪い効果の主な制限が存在します。注入された受精卵の着床後に生まれた動物の数パーセントしか遺伝子を統合しています。対照的に、レンチウイルスベクターは統合的な遺伝子導入のための強力なツールをヘッジホッグ受精卵の使用により、創設者の高効率生産 70% 以上の平均収量を持つトランスジェニック マウス。さらに、遺伝子組換え動物を生成する任意のマウスの緊張を使用ことができますおよび遺伝子発現の浸透度がきわめて高く、DNA マイクロインジェクションに比べてレンチウイルスベクターを介した遺伝子導入で 80% 以上。レンチウイルスベクターによって貨物をすることができます DNA のフラグメントのサイズは 10 kb に制限されている、このメソッドの主要な制限を表します。シンプルで簡単に受精卵の透明帯の下に注入の手順を実行を使用して、50 以上の創設者動物はマイクロインジェクションの単一のセッションで生成できます。このようなメソッドは、創設者動物、急速な利得と損失関数学または画面ゲノム DNA 領域の制御および遺伝子発現生体内で調整する機能のために直接実行する高い適応です。
概要
1980 年にゴードンらの先駆的な仕事を示した偽妊娠マウスに着床した受精卵前核の男性のにプラスミド DNA 注入は統合トランスジェニック動物の生産をもたらすことができる、プラスミド DNA の1。トランスジェニック哺乳類が生成されることを実証では、両方の基礎科学と医療薬学研究の新規分野への道を開く、ライフ サイエンスの世界に大きな影響を与えた。過去 4 年の間、DNA マイクロインジェクション ルーチンの練習となっています。トランスジェニック マウスの膨大な数が生産されている、標準的な方法すべてのマウス系統の完全に使用可能ではないと時間のかかる戻交配2,3が必要です。他の種への応用のまま挑戦4全体の transgene の統合収量生まれた動物5の数の割合に制限されます。また、transgene の統合の効果は、前核 DNA インジェクションの貧しい全体的な利回りを説明する制限要因を表します。この点で、統合ウイルスベクター貨物に最も効率的なツールし遺伝子を統合し、こうして大幅統合収穫、遺伝子サイズ 10 kb6 を超えることのできないという唯一の制限を増加する新しい手段を提供できます。.
レンチウイルスベクター擬似エンベロープ蛋白の水疱性口内炎ウイルス (VSV) 型指定された pantropic および高度統合遺伝子伝達ツール、受精卵7を変換する使用ことができます。卵子の周囲の透明帯自然ウイルス障害とレンチ ウイルスのベクトルを伝達できるように渡される必要があります。トランスジェニック動物はマイクロ ドリルや透明帯8,9の削除後変換受精卵母細胞によって生成されています。ただし、囲卵腔に透明帯の下に注入はロイスと同僚の7で最初に定義されている受精卵を変換する最も簡単な方法のように見えます。
レンチウイルスベクターの囲卵腔注入では、生まれた動物の 70% 以上は、トランスジェニック動物の生産に高収率をことができます。このような利回りは標準的な前核DNA 注入7,10,11を使用して達成することができます最高の収量より 10 倍以上高い。この文脈では、注射の 1 つのセッションは少なくとも 50 トランスジェニック創設者 (F0) を生成します。創設者の多数は、したがって、トランスジェニック マウスの行を生成することがなく F0 マウスに対して直接遺伝子効果の表現と互換性があります。この利点は transgene 効果の迅速なスクリーニングを可能、週間以内に体内の利得と損失関数の研究を実行に特化。また、エンハンサーと転写因子11,12によってバインドされている DNA モチーフをマップする調節 DNA 配列することができます急速に上映されるも。前核の注射で遺伝子は通常ユニークな軌跡の複数コピーを統合します。レンチ ウイルスのベクトル統合は軌跡10,13あたり単一コピーとして多遺伝子座で発生します。したがって、統合遺伝子座の多様性より堅牢な新しい生成モデルを作るトランスジェニック創設者にみられる非常に高い式浸透に関連付けられている可能性が最も高いです。
重要なは、DNA の前核の注入を使用して、プロシージャの間に前核の可視化は絶対に必要があります。この技術的な制限は、多種多様なマウス系統から受精卵の使用を防止します。したがって、特定のトランスジェニック モデルの生産目的のマウスの遺伝子を転送する少なくとも 10 連続戻交配続いて寛容ひずみにおける動物の生産を要求するどの前核が表示されないためにのひずみひずみ。レンチウイルスベクター注射囲卵腔が表示常に、注射は非常に特定のスキルを必要はありません。例として、前核注入に適さない NOD/SCID トランスジェニック マウス ウイルスベクター注射14得られています。
ここでは、包括的なプロトコルが 1 つのセル段階の胚の囲卵腔注射レンチウイルスベクターを用いた遺伝子改変マウスの簡単な生産を許可するように表示されます。遺伝子発現のいずれかで制御ユビキタスまたはセル特定のプロモーターで詳しく説明します。
PTrip ΔU3 レンチ ウイルスのバックボーンは、この研究は15で使用されました。このベクター U3 シーケンスを U3 プロモーター活性を外し、自己不活化ベクトル (罪)16を生成する部分的に削除されているレプリケーション欠陥レンチウイルスベクターを作り出すことができます。P8.91 カプセル化プラスミド (ΔVpr ΔVif ΔVpu ΔNef)6、水胞性口炎ウイルス (VSV) 糖 G17、および pTRIP ΔU3 のエンコーディング pHCMV G と HEK 293 t 細胞のトランスフェクションによって生産されたレンチウイルスベクター株式組換えのベクトル。詳細な運用手順は、補足的なメソッドとして提供されます。
高価レンチウイルスベクターの在庫の生産、バイオ セーフティ レベル II 条件 (BSL-2) の下で実行されます。これは BSL 3 で生産されなければならない遺伝子を除いてほとんど transgenes に当てはまります。したがって、ほとんどの場合のための BSL 2 条件での生産は十分です。さらに、使用と生産は、遺伝子組み換え作物 (GMO) を扱うほとんどの国の規制当局の切断されます通常。(P24 キャプシド蛋白質の 2 μ g) の下に複製無能な罪レンチウイルスベクターの限られた量は、欧州連合推奨事項に従ってフランス GMO 代理店によって前述の BSL 1 条件下で使用できます。
プロトコル
動物の仕事を含むすべての手順は、倫理的な承認を受けたし、フランス研究番号 APAFIS #5094 20 16032916219274 v6 の下教育省と 05311.02 によって承認されています。PHENOPARC は、フランス農水省認定番号 B75 の下で認定されている ICM 動物施設 13 19。総合的なプロトコルでは、図1 にまとめられている正確な時間枠内で各手順を実行する必要があります。
1. 動物の購入および基本的な化合物の合成
- 動物の購入
- 25 精管男性 B6CBAF1/JRj は 8 週齢 (♀C57Bl/6JRj、♂CBA/JRj の間元から F1 世代を越える) を注文します。
注: は、到着時に男性を分離します。男性の精管は少なくとも 1 年間再利用できます。
ケージを 3 週間ごとに変更します。 - 10 週齢は、少なくとも 50 の動物のプールを保持する 50 の B6CBAF1/JRj 女性を注文します。
- 8 週齢は、10-15 C57BL/6JRj の肥沃な男性を注文します。
- 4 週齢は、30 の C57BL/6JRj 女性を注文します。
- 25 精管男性 B6CBAF1/JRj は 8 週齢 (♀C57Bl/6JRj、♂CBA/JRj の間元から F1 世代を越える) を注文します。
- 麻酔および安楽死
- 300μL ケタミン ・ キシラジン ミックス、腹腔内注入量を用いて麻酔 (150μg/g 体重、0.15μg の用量でキシラジンの用量でケタミン g 本体重量/)。動物は、体の温度を調整するパッドを加熱下で配置されます。手順を開始する前に、動物の尻尾をつまんで反射神経をチェックします。
- 安楽死は、頚部転位によって行われました。斬首は、動物の死を確認する第 2 の方法として含まれていた。胚の安楽死は、斬首によって行われました。
注意: 到着時に許可動物施設 (処理や交尾) に習慣になって 1 週間以上。重要なは、遺伝子組換えの行を含む任意のマウス系統は、肥沃な男性と排卵の肥沃な女性として利用できます。ひずみの選択は、科学的な質問の要件によるとすべきであります。
- ホルモン製剤
- 1 凍結乾燥 PMSG バイアルに PSMG (妊娠馬血清ゴナドトロピン) バッファーの 910 μ L を追加、100 μ 因数を作る、-20 ° C で保存
注: 各因数には 11 マウス用 55 UI が含まれています。最初の使用後 2 週間以上、PMSG 因数を決してしてください。 - 2730 μ L 1 hCG (ひと絨毛性ゴナドトロピン) バッファーの凍結乾燥バイアル hCG を追加します。100 μ 因数をし、-20 ° C で保存
注: 各因数には 11 マウス用 55 UI が含まれています。
- 1 凍結乾燥 PMSG バイアルに PSMG (妊娠馬血清ゴナドトロピン) バッファーの 910 μ L を追加、100 μ 因数を作る、-20 ° C で保存
- ヒアルロニダーゼ準備
- ヒアルロニダーゼの M2 10 mg/mL の原液を入手し、50 μ 因数を中長期の 3 ml のバイアル 1 を再構築します。その後、-20 ° C で保存します。
- 手術ツール準備
- オートクレーブを使用してすべての手術ツールを滅菌します。
2. 女性ドナーの排卵
- 12 h を使用して動物施設におけるデイ - 夜サイクル 14-3 日で PMSG を注入します。HCG 注射の直後に日-1 とチームメイトの午前 12 肥沃な男性で hCG を挿入します。
- PMSG の 100 μ L の 1 因数に無菌の 0.9 %nacl 溶液の 1 mL を追加-3 日 (55 UI)。腹腔内任意のデッド ボリュームがない注射器を使用して 100 μ L で C57BL/6JRj 女性 10 名を挿入します。
注: 各マウス PMSG の 5 の UI が表示されます。 - -1 日に hCG の 100 μ L の 1 因数に無菌の 0.9 %nacl 溶液の 1 mL を追加 (55 UI)。腹腔薄めた hCG (5UI) の 100 μ L で PMSG 注射を受けたマウスを挿入します。任意のデッド ボリュームがない注射器を使用します。ゆっくりと注入を実行し、液体が漏洩しないように、針を削除する前に待機します。
注: 各マウス hCG の 5 の UI が表示されます。HCG の注射は PMSG 後 46 h を行わなければなりません。 - HCG 注射後直接スタッド オスのケージに各 C57BL/6JRj の女性を配置します。
- 0 日目の朝に膣にプラグを確認し、受精卵を収集するために肯定的な女性を使ってください。
3. B6CBAF1/jRj 偽妊娠女性の準備します。
- 採卵 (日-1) 2 B6CBAF1/JRj 女性と 17 で前日精管男性を仲間します。
注: 女性のサイクルの同期を回避する別のケージから起きている女性でメイトに非常に重要です。これは膣にプラグを得る収量を高めます。さらに、過去 2 日以内に変更されているオスのケージにメスを追加しません。ケージが汚い場合、雄の繁殖行動の有効性が高い。
4. 受精卵コレクション
- 準備
- 1450 μ M2 をヒアルロニダーゼ作業ソリューションを準備するヒアルロニダーゼ原液に追加します。
- 100 mm シャーレで受精卵を生産し、室温で維持するために使用女性あたりヒアルロニダーゼ実用的なソリューションの 100 μ L の一滴を配置します。
- 4 井戸のプレートに M16 の 500 μ L を追加します。2 井戸注入するレンチウイルスベクターの種類ごとを使用: 1 つも挿入された卵とその他が含まれます非注入されたもの。5% CO2の大気中の 37 ° C の定温器に 4 のウェル プレートを配置します。
- 胚の採取及び取扱のピペットを準備します。
- 炎の硬質ガラス キャピラリー チューブの中心を回転させることによりガラス ヘマトクリット管 (75 mm/60 μ L) を柔らかきます。
- 可能であり、管の約 300 μ m プルきちんと休憩を取得する冷却管の内部の直径を取得するプルとしてすぐに熱から毛細血管を削除します。
- 卵管を収集します。
- 0 日 9 で頚部転位による C57BL/6JRj 女性を安楽死させます。死は、斬首によって確認されます。
注: この安楽死法、IACUC によって承認された、欧州の推奨事項に従います。 - 大きな水平切開をハサミで腹腔内を開くを実行します。卵管は子宮と卵巣の間に位置します。
- Mesometrium とカーブタイプ鉗子で著名な血管を運ぶ膜を削除します。
- カーブタイプ鉗子で卵巣から卵管を区切ります。
- 曲線はさみを使用して卵巣から卵管をカットするのにガイドとしてカーブタイプ鉗子を使用します。
- 卵管を引き、子宮から曲線はさみでカットします。
注意: は、受精卵が含まれて腫れた乳頭を触れないでください。滅菌器具を使用して全体の手順を実行します。 - M2 培地 (35 mm ディッシュ) 室温で収集したすべての卵管を配置します。
- 同じ 100 μ L ヒアルロニダーゼ作業ソリューション (0.3 mg/mL) 中 2 卵管を配置します。
- 0 日 9 で頚部転位による C57BL/6JRj 女性を安楽死させます。死は、斬首によって確認されます。
- 受精卵から卵丘細胞を削除します。
- 2 インスリンは注射器を使用して、顕微鏡の下で: ヒアルロニダーゼ作業ソリューションに卵管を保持するために最初の 1 つと 2 つ目、乳頭を引き裂くし、分散する受精卵。
- 卵を収集するために準備ガラス ピペットを取るし、チューブと 0.22 μ m のフィルターのすべての卵を吸引するマウスピースのマウントに接続します。すべての卵を収集し、連続通過で M2 媒体の 100 μ L の 6 滴、別に洗ってください。
- 場所は、M16 中 5% CO2の大気と加湿の 37 ° C の定温器に受精卵を洗浄しました。
5. 作るインジェクション ピペット
- 外径 1 mm の薄肉ガラス キャピラリー チューブ (長さ 10-15 cm) を使用して、水平方向のピペットの引き手に毛細血管をクランプします。
注: 最も水平ピペット引き手、3 つのパラメーターことができます調整する: 熱量、加熱と引きの強さと時間遅延を引っ張るします。図 2 aで 1 つのような注入の pipets を取得するこれらのパラメーターを調整します。DNA マイクロインジェクションを日常的に実行しているユーザーは、標準の設定を使用して、暖房と引っ張ってピペット チップのグローバルの形状を変更する間の遅延を調整する」を参照してください。
6. ピペット持株を作る
- 注入のピペットを使用して、ホールディング ピペットを準備します。
- マイクロフォージに注入するピペットを取り付けます。80 を 100 μ m 径の先端の鋭利な対称を取得するマイクロフォージとピペットをカットします。マイクロフォージ鋭いヘッジなしの対称的なラウンド形状を得るために熱のヒントをポーランド語します。
7. レンチ ウイルス ベクターを含むインジェクション ピペットの準備
- 冷凍レンチ ウイルス株によくある破片をペレットに 2 分間、160 x g のレンチウイルスベクター懸濁液を遠心します。
- 上清を回復し、クラス II の安全キャビネットで新しい 0.5 mL の管にそれを転送します。
- 5 マイクロ ローダーを使用しての手順で説明されているインジェクション ピペットに 1 μ L の上清を転送します。
- 右のマイクロマニピュレーターの計測器ホルダーにインジェクション ピペットを設定します。ホールディング ピペットを左のマイクロマニピュレーターに接続します。
注: のレンチウイルスベクターの伝達力価は、創始者生産の有効性に直接関連付けが。高効率のため (> 70%)、ウイルスのベクトルを使用して、100 の範囲の価と p24 キャプシド蛋白質/μ L の ng。価が 10 を超えるべき価伝達単位 (TU) で表す場合、9 TU/mL。レンチウイルスベクター株式は p8.91 試験管プラスミドと 293 t 細胞のトランスフェクションによって生成される必要があります pHCMV G、補足方法18で説明されているように水胞性口炎ウイルス (VSV) の糖蛋白質 G をエンコードします。
8. マイクロ射出成形
- 光パラフィン オイル (胚テスト) M2 中うつ病スライド カバーの中心の 8 μ L を分注蒸発を避けるために。
- 場所 20 卵にできるだけ分散した少なくともとしてドロップ。
注意: 胚を沈殿させた場合は、泡を行いません。 - インジェクション ピペットの先端が開いていることを確認します。されていない場合は、ホールディング ピペットでインジェクション ピペットをタップします。
- 20 の噴射時間のガラスシリンジを設定 s。
メモ: ウイルスの懸濁液の粘度は、囲卵腔にウイルスのベクトルの分散の明確に可視化できます。射出圧力は 20 内全体のスペースを埋めるために調整する必要があります, 10 ~ 100 の容積を表す噴射の s の Pl射出圧力が 600 hPa を超えない。 - 2 プロ核と、実体顕微鏡下でホールディング ピペットで 2 極体が含まれている 1 つの受精卵を吸引します。
- マイクロインジェクター 8.4、囲卵腔内に記載されている設定を使用して、卵を注入します。
注意: は、インジェクション ピペットで細胞膜を触れないでください。 - 20 卵のバッチで利用可能なすべて受精卵を注入し、5% CO2の大気と加湿の 37 ° C の定温器に事前に加熱された M16 中すぐに注入された卵を配置します。
注: は、胚の転送前に注入後 30 分以上の挿入された卵を孵化します。
9. B6CBAF1/JRj 偽妊娠の女性に胚を転送します。
- B6CBAF1/JRj パイプカット男性と B6CBAF1/JRj 雌を交尾後交尾プラグ 16 h を確認します。ちょうど卵のコレクションを開始する前にこれを行います。
- 注入された胚の注入のピペットを準備します。
- 前述の収集および処理 (手順 4.2) の胚の胚の注入のピペットを作る。長さで約 4-5 cm の狭い部分で約 150 μ m の内径とピペットを選択します。
注: 先端は炎卵や卵管に損傷を減らすために研磨をする必要があります。- ピペットの肩のすぐ上光パラフィン オイル (胚テスト) と入力します。
- 小さな気泡、M2 の中規模、および最終的に第 2 空気の泡を吸引します。
- 胚を胚と一緒に卵管に注入される媒体の容量を最小限に抑えるために互いの背後にある 1 つを作成します。
- 約 1 つの胚の幅の光パラフィン オイル (胚テスト) の非常に小さなドロップをロードすることによって終了します。
注意: は、ピペットを処理しながら、優しくすること。
- 前述の収集および処理 (手順 4.2) の胚の胚の注入のピペットを作る。長さで約 4-5 cm の狭い部分で約 150 μ m の内径とピペットを選択します。
- 胚移植。
- すべての器具を滅菌します。
- 滅菌 0.9% 食塩のケタミンの体重 g あたり 150 μ g とキシラジンの体重 g あたり 0.15 μ g を含む 300 μ の腹腔内注入による女性を麻酔します。
- 動物の尻尾を鉗子でつまんで麻酔深度を確認し、subcutaneouly 0.1 mg/kg の前にプロシージャを開始 (ブプレノルフィン) 鎮痛剤を注入します。
- 最後の肋骨のレベルで脊髄に沿って背中の両側に 2 cm を剃る。
- 加熱パッドや滅菌フィールドでは、女性のマウスを置きます。マウス背面の中央に 2 cm × 2 cm のウィンドウをカットします。
- 皮膚の消毒液 (10% ポビドン ヨウ化) を適用し、はさみで 1 cm の横切開を作る卵巣 (オレンジ色) が体壁を介して表示されるまで皮膚を横方向にスライドします。
- 両眼手術顕微鏡下で高級ハサミで体壁卵巣のすぐ上に 5 mm の切開を確認します。
- 非外科的ブルドッグ留め具付き脂肪パッドをピックアップし、卵巣、卵管、子宮の上部を引き出します。
- 膨大部を視覚化し、膨大部に卵巣をリンクしている卵管セグメント上 vannas ハサミ、ヘミセクションを作る。
- 転送胚ピペットを紹介し、注入ピペットで最初の空気の泡で停止、膨大部に卵をお届けします。
- 2 回目の卵管に手順を繰り返します。
- 傷クリップで皮膚をクローズ アップ。
- 場所回復で動物の部屋 (39 ° C、30-60 分) まで完全に目を覚まし。
- 鎮痛剤注射 12 時間後、痛みや苦痛の兆候の場合 48 h を繰り返します。
- 手術後 7-10 日傷のクリップを取り外します。
- 注入後は 3 日おき、ウェイト カーブに従うことによって注入された女性の妊娠を確認してください。重要な体重増加は注入後 10 〜 12 日間を観察することができ、妊娠を示すものになります。
注: ここでは今後すべての胚推定トランスジェニック創設者を表しています。これらの創設者の表現型は、すべての発達段階でまたはこれらのトランスジェニック動物の世代にリンクされた科学的な質問によると出生後に分析できます。
10. ジェノタイピング トランスジェニック創設者
- 10 ミリメートル pH 8; トリス塩酸を含む遺伝子型バッファーを準備します。5 ミリメートルの EDTA、pH 8.0 0.2 %sds (w/v)、50 mM NaCl。0.22 μ m のフィルターを通してジェノタイピング バッファーを殺菌し、数ヶ月常温で保存します。
- (胚) のために初期胚胚体外膜または 500 μ L に (生まれた動物) の尾の小さな作品ジェノタイピング バッファーをフィルター処理し、プロティナーゼ K の 15 μ L (20 mg/mL) を追加します。55 ° C で一晩インキュベートします。
- ライセート 5 分 15,000 × g で遠心し、PCR 反応の清 1 μ L を使用します。ライセートは、数ヶ月 4 ° C で格納することができます。
- 1 x PCR バッファー、1.5 mM MgCl2、各消化サンプルの 1 μ L、1 UI の Taq DNA ポリメラーゼ 0.2 μ M 各 PCR のプライマーの dNTPs の 200 μ M を含む 20 μ L 反応量の遺伝子断片の PCR 増幅を実行します。ネガティブ コントロールとして使用 1 μ H2o.肯定的な制御として遺伝子を含んでいるレンチウイルスベクター プラスミッドの DNA を使用します。
- EGFP の検出のためには、使用を説明します。
eGFP 前方プライマー: 5' 3' GACCACATGAAGCAGCACGACTTCT
eGFP 逆プライマー: 5' 3' TTCTGCTGGTAGTGGTCGGCGAGCT - たちで PCR 増幅を実行: 94 ° c、60 ° C で 1 分 1 分の 35 サイクルに続いて 94 ° C で 4 分、72 ° c. で 2 分
- 図 2 bに示すように 300 bp eGFP PCR 製品を視覚化するために 2% の agarose のゲルの PCR の製品をロードします。
注: 300 bp PCR バンドを示すすべての個人は eGFP 遺伝子を統合している、遺伝子組み換えとして見なすことができます。 - トランスジェニック動物の遺伝子発現を分析します。たとえば、実行組織として染色図 3と図 4に示す補足の方法で説明し、。
注: 両方地球科学的問題に応じて適切な方法を使用して、遺伝子発現解析と表現型解析を実行する必要があります。
11. 遺伝子コピー数の定量
- 量的な PCR のための DNA のサンプルを準備します。
- 種の DNA 抽出 (gDNA) プロティナーゼ K ライセートから 10.3 の製造元の指示に従って商業キットを使用しての手順で取得します。
- 260 吸光光度法による gDNA を定量化する nm。
- 各 gDNA サンプル 10 ng/μ L の最終濃度を薄くしなさい。
- 各サンプルの準備 5 シリアル希薄 (1:5) で H2O 以下の濃度で 6 チューブを取得する: 10 ng/μ L、2 ng/μ L、0.4 ng/μ L、0.08 ng/μ L、0.016 ng/μ L、0.0032 ng/μ L
- 定量的 PCR (qPCR) 反応混合物を準備します。
- QPCR のプライマー ミックスを準備します。QPCR 用それぞれのプライマーのカップル、前方プライマー (100 μ M プライマー原液) を 10 μ l 添加、逆プライマー (100 μ M) の 10 μ L を追加し、80 μ H2o.
注: eGFP を増幅、TCCAGGAGCGCACCATCTTCTTCA 前方、逆 TTGATGCCGTTCTTCTGCTTGTCG のプライマーを使用します。Cdx2遺伝子は、qPCR (ゲノムごとの 2 つのコピー) のノーマライザーとして使用されます。Cdx2ノーマライザー GCCAGGGACTATTCAAACTACAGG 前方、逆 GACTTCGGTCAGTCCAGCTATCTT のプライマーを使用します。 - EGFP プライマー ミックスと Cdx2 プライマー ミックスの 2 qPCR ミックスを準備します。各 gDNA の重複 6 希釈液を増幅するための十分な qPCR ミックスを準備します。QPCR 反応 1 つにはには、3.8 μ H2O、x 反作用の組合せ蛍光緑 2 の 5 μ L とプライマー ミックスの 0.2 μ L が含まれています。
注:、テストする各のトランスジェニック動物の 24 の qPCR 反応を行います。反応混合物は、384 よく qPCR 用に提供しています。 - それぞれの動物テスト、配布: Cdx2 qPCR ミックスの 9 μ L eGFP qPCR ミックス ・ 12 井戸 9 μ L と 12 の井戸。EGFP qPCR ミックスを含む 2 井戸と eGFP qPCR ミックスを含む 2 の井戸に各 gDNA 希釈の 1 μ L を追加します。
- 各 qPCR の 2 のまま井戸ミックスどの gDNA を H2O ネガティブ コントロールとして交換されました。
- QPCR のプライマー ミックスを準備します。QPCR 用それぞれのプライマーのカップル、前方プライマー (100 μ M プライマー原液) を 10 μ l 添加、逆プライマー (100 μ M) の 10 μ L を追加し、80 μ H2o.
- QPCR マシンに 384 ウェル プレートを配置し、次の実行しているプロトコルを適用: 95 ° C、50 で 10 分サイクル、10 95 ° C および 60 ° C で 1 分で s
- データを分析します。各 gDNA をテストするには、Ct 値を合計 gDNA 量 (重複の 6 ポイント) のログの関数としてプロットします。最小二乗法で線形回帰を使用してカーブに合わせて、y 軸の切片に対応する Ct 値を推定します。Cdx2 基準 eGFP コピー数を計算する eGFP と正規化された Cdx2 の推定の Ct 値を使用 (2 のコピー) 標準的な 2ΔdCt法11を使用しています。
結果
トランスジェニック動物は、ここで提示されたプロトコルを使用して生成されました。代表の結果両方のユビキタスと細胞タイプ特定の遺伝子発現を示します。
導入遺伝子の発現
ユビキタスのプロモーターは、持続的かつ効率的な方法で遺伝子を表現する基本的な研究ツールです。このようなプロモーターは、小規模および大規模な動物の生体内での遺伝子導入細胞の in vitroトランスフェクションからアプリケーションの非常に大きい変化に使用されます。
レンチウイルスベクターは、サイトメガロ ウイルス (CMV) プロモーターまたは CAG 鶏アクチン プロモーターと CMV エンハンサーの融合に基づく複合プロモーターの制御下で緑色蛍光レポーターの遺伝子 (eGFP) を表現するために建設されました。両方のレンチウイルスベクター生産 (補足メソッド)、力価は eGFP 発現に基づく伝達単位 (TU) として 293 t 細胞で決定されました。レンチウイルスベクターの構造化は、109 TU/mL の注入で擬似妊娠したメスマウス濃度で受精卵母細胞の囲卵腔に注入されました。EGFP 統合に従う PCR によって次に移植胚が出産直前に収集され、データ。73% (n = 22) 83% (n = 32) 収集した胚の統合した遺伝子、CMV と CAG レンチ ウイルス構造のそれぞれ (表 1)。トランスジェニック胚は、免疫染色 eGFP をしていた。CAG プロモーターを使用した場合、すべてのセルは GFP を表明したに対し、散乱 eGFP 陽性細胞のみを CMV プロモーター (図 3、上部パネル) と観察図 3に示すように、(図 3、中間と下部のパネル)。
CAG プロモーターと収集したトランスジェニック胚の 96% は遍在 eGFP 遺伝子 (表 1) を表明しました。両方のプロモーターは、ユビキタス、CAG プロモーターだけはドライブすべての細胞で遺伝子の堅牢な表現することができます。代替ユビキタス プロモーターはホスホ グリセリン酸キナーゼ (PGK) とユビキチン C プロモーターなどに使用され、eGFP (データは示されていない) の発現レベルが低い CAG プロモーターで得られるものと同様の結果が得られました。
生体内でティッシュ特定のコントロール要素をテストする規定するゲノム領域のマッピングします。
多数のアプリケーションでは、トランスジェニック動物の細胞特定の方法で遺伝子の発現が必要です。さらに、トランスジェニック動物の世代は、画面の特定の遺伝子の細胞特異的発現を制御する推定の規制のゲノムの DNA のフラグメントの能力に非常に貢献することができます。たとえば、トランスジェニック動物のレンチウイルスベクターを介した生産はニューロジェニン 3 (Neurog3) 式11を制御する細胞特定のエンハンサーをマップする使用されました。Neurog3膵前駆細胞内分泌の運命へのコミットメントを制御基礎螺旋形ループ螺旋形 (bHLH) トランスクリプション要因であります。Neurog3 null 変異マウスの膵臓の内分泌細胞ない19を区別できます。前述したようにドライブ、eGFP 遺伝子発現に β グロビン最小プロモーター レンチ ウイルスのベクトルの上流に位置-5284 とサイトを開始Neurog3転写に対する-3061 間ローカライズ 2.2 kb DNA 断片がクローン11。 制御構造は同様に 6 マウス染色体上に局在した 2.4 kb のコード断片をクローニングによって生成された (chr6: mm9 マウスのゲノムのアセンブリを基準と 14237279 14239685) 同じレンチ ウイルス バックボーン。このゲノムの領域は、 Gpr85とPpp1r3a遺伝子間 1 メガ ベース長い遺伝子砂漠内でローカライズされます。高価レンチウイルスベクターは次構造と名前付きNeurog3- ガ-eGFP と Chr6 eGFP 両方を使用して作り出されました。
両方のレンチウイルスベクターを構築し、(補足メソッド) を生産します。現在利用可能なNeurog3を発現する細胞がないので TU 価を特定できませんでした。また、p24 キャプシド蛋白質の濃度としては価を測定しました。二つのベクトルは受精卵母細胞の囲卵腔内に注入され、擬似妊娠雌マウスに移植します。この発達の段階が膵臓のNeurog3の最大の式に対応して、移植胚を胚日 14.5 (E14.5) で採取。胚は eGFP 統合に従う誘因次.84% (n = 47) と 71% (n = 48) 収集した胚の統合したNeurog3- ガ-eGFP と Chr6 eGFP レンチ ウイルス構造遺伝子それぞれ (表 1)。各胚の膵臓の芽を切開し、免疫染色を行う断面します。Neurog3- ガ eGFP トランスジェニック胚の 92% は、図 4上部パネル (代表的な染色) に示すように膵臓の eGFP を表明しました。重要なは、eGFP 陽性細胞も 2.2 kb Neurog3エンハンサーを示す Neurog3 発現細胞 (図 4) の大半は eGFP 発現Neurog3セル人口を制限することができます。野党、Chr6 eGFP 胚のどれもは eGFP (図 4の下部のパネルおよび表 1) 膵臓や膵臓の外を表明しませんでした。また、eGFP の異所性発現が認められなかったNeurog3- ガ eGFP 胚11膵臓の外。
上記 4 実験の手順の各ステップの正確な定量的説明は表 1に示したが。これは、プロシージャの世界的な有効性を示しています。確かに、番号注入受精卵に遺伝子を統合されて収集した動物の数を比較すると、プロシージャのグローバルの収量は平均で 44%。Β-ガラクトシダーゼ レポーターに融合 Neurog3 エンハンサーを含むコンストラクトの前核 DNA 注入で同じ収穫量は 3.1% を超えない。
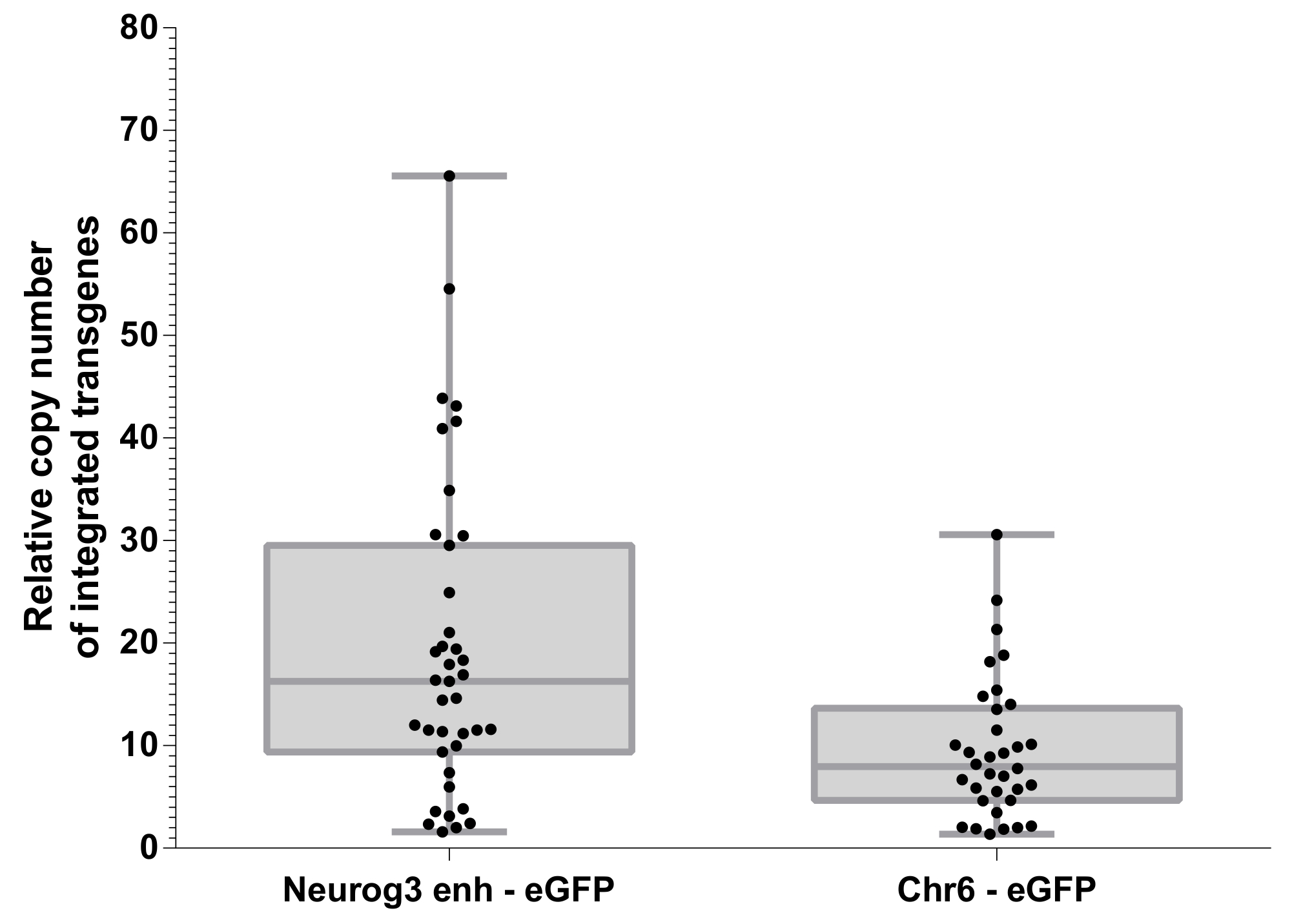
レンチ ウイルスのベクトルを持つ受精卵母細胞の情報伝達は、複数サイト10,13で発生することができます transgene の統合に します。Transgene の統合サイトの相対数は、ゲノム DNA (図 5) の量的な PCR を用いて評価しました。EGFP 統合の定量化された量的な PCR (qPCR) によって決定され、11を前述のようにゲノムあたり 2 コピーであるCdx2遺伝子に正規化されました。統合サイトの平均数は 19.36 ± 2.468 (S.E.M.) とそれぞれNeurog3- ガ-eGFP と Chr6 eGFP コンス トラクターから生成された胚の 9.537 ± 1.186 (S.E.M.) だった。興味深いことに、これらの動物を生成するために使用 2 つのレンチウイルスベクターは別ウイルス抗体価を発表しました。P24 キャプシド蛋白質の濃度はNeurog3- ガ eGFP ベクトルの 124 ng/μ L のと 52 ng/μ L Chr6 eGFP ベクトル制御のための.トランスジェニック胚 (図 5) の両方の人口に統合サイト番号において有意差をそのような力価の差が占める可能性が最も高いです。これは、創始者トランスジェニック マウスのバッチで得られる統合サイトの平均数を異なる抗体ウイルス株を用いた変調可能性を示唆しています。
重要なは、直接的な相関関係が認められなかったNeurog3- ガ eGFP トランスジェニック胚における eGFP の発現と統合されていた遺伝子のコピーの数。つまり、統合Neurog3- ガ eGFP 遺伝子のコピーを 1 つまたは複数の胚は同様に、Neurog3 陽性細胞のエクスプレス eGFP と見つけられました。

図 1: 全体の手順のフロー図この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}

図 2: マイクロインジェクション ピペットと遺伝子型の準備。
(A) DNA やレンチウイルスベクター注射用 pipets の主な違いを強調するマイクロインジェクション ピペットの模式図。左右パネル: 両方のピペット タイプの全体的な形状を描画します。破線の円は、ピペット先端部の拡大領域を強調表示します。ピペット チップの写真が掲載されてもいます。先端はレンチ ウイルス注射用、壊れた点線と対応する画像で示される必要があるということに注意してください。右側のパネル:卵注射のいずれかの 1 つの前核の左、受精卵、インジェクション ピペット ホールディング ピペットで設定または囲卵腔での例。スケール バー = 50 μ m eGFP PCR の製品の agarose のゲルの(B)の可視化 8 異なる胚 (1 に 8 車線) から抽出したゲノム DNA からの増幅します。唯一の胚 1、2、3、5、6、8 は eGFP 遺伝子を統合しました。DNA プラスミド pTrip PGK eGFP レンチウイルスベクターの生産に使用される PCR 陽性対照として使用されました。陰性対照の H2O は PCR の反作用の DNA を交換しました。MWM: 分子量マーカーです。bp = 塩基対。この図の拡大版を表示するのにはここをクリックしてください。
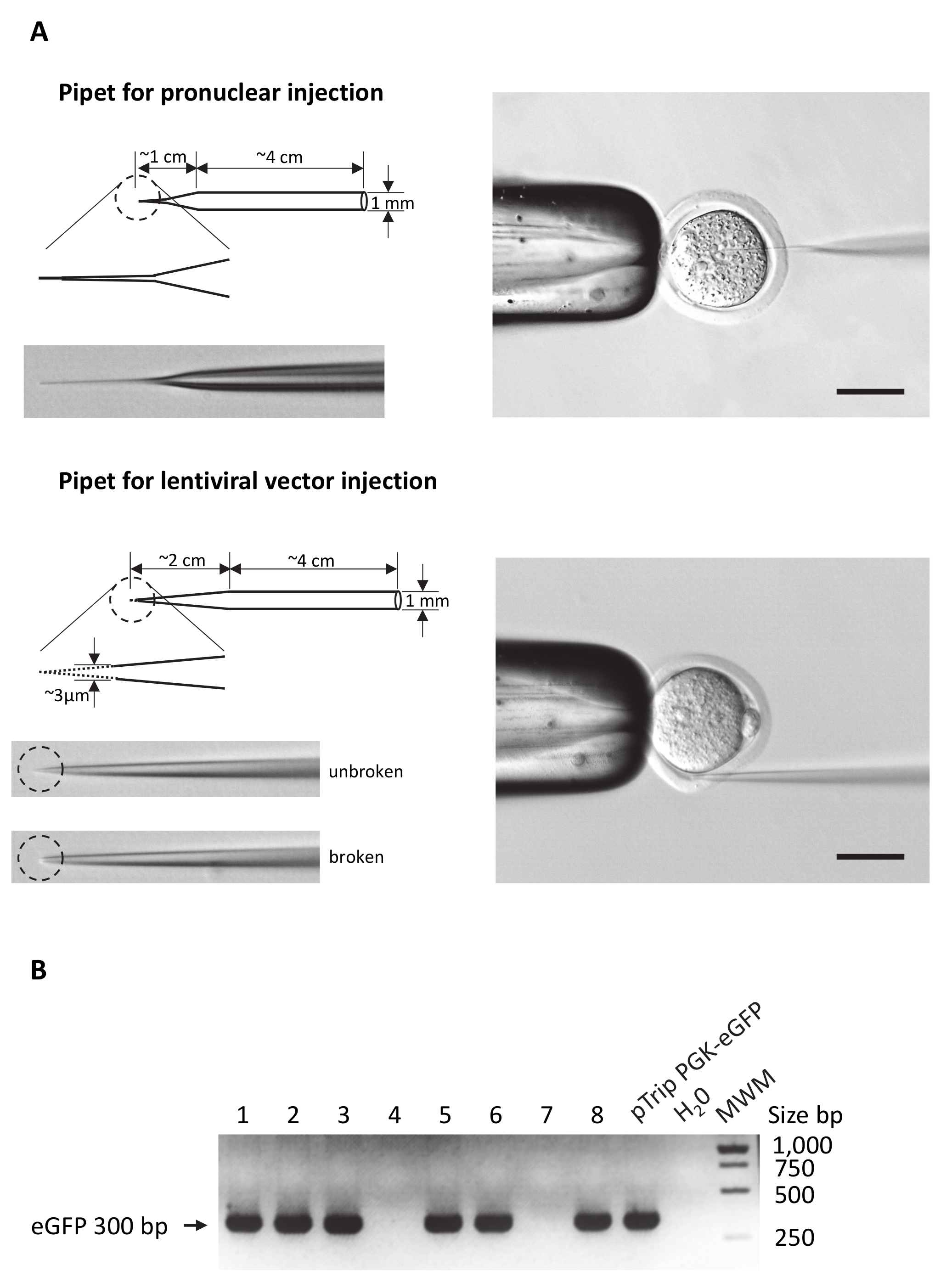{kind=link}

図 3: ユビキタス プロモーター遺伝子胚における eGFP レポーターの発現をドライブします。
10 μ m 低温 - トランスジェニック胚の染色 eGFP 発現 (緑) と (青) の核を視覚化します。レンチ ウイルス CMV プロモーターで生成された胚を構築 (上左ラベル) が E11.5 で収集されました。E18.5 で CAG プロモーター レンチ ウイルス構造 (下部左ラベル) で生成された胚を採取されました。Pb: 膵芽、 VSC: 腹側脊髄、 Vt: 椎、李: 肝臓、Ms: 腹部のベルトの筋肉。スケール バー 50 μ m =この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}

図 4: トランスジェニック胚におけるレポーター遺伝子の細胞特異的発現Neurog3エンハンサーによって駆動されます。10 μ m 低温-形質転換胚の E14.5 膵芽の染色補足的な方法に記載されている Neurog3 (赤), eGFP (緑) と核 (青) の発現を可視化する)。Neurog3 式は、膵臓内に散在しています。統合Neurog3- ガ-eGFP トランスジェニック胚作成エクスプレス eGFP と eGFP 陽性細胞のほとんどは、Neurog3 陽性 (上部パネル)。胚 Chr6 eGFP コンス トラクターで生成された eGFP (底板) を表現していません。スケール バー 50 μ m =この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}

図 5: 統合された遺伝子の相対的なコピー数。CDX2 遺伝子プロトコル セクションで説明した基準 eGFP 統合サイトの定量化。75 パーセンタイルに 25thからボックス プロット。ドットは、生成された異なる遺伝子胚を表しています。2 つのレンチ ウイルス構造間の transgenes 統合サイトの比較が大幅に異なる (不対パラメトリック t 検定, p = 0.001)。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}

表 1: 手順中に一歩一歩定量的レポート。手順の中に、卵や胚の数が数えられました。最初の列は、排卵の雌の卵管から取得された卵の総数を表します。2 極体をオフにのみ卵および/または表示前核を注入したし、報告されます。注入および文化で数時間後なくを溶解し、正常な形態を持っていた注入された卵のみを移植されました。次に偽妊娠の女性から採取した胚の合計数がカウントされます。最後に、遺伝子を統合し、記者を表明した胚は、最後の 2 つの列に表示されます。同じ機能は、標準的な前核 DNA 注入を用いた実験で比較のためまた与えられます。ここで遺伝子には、β-ガラクトシダーゼ レポーター遺伝子 (Neurog3- ガ-LacZ) の発現を運転Neurog3エンハンサーが含まれています。このファイルをダウンロードするここをクリックしてください。
ディスカッション
レンチウイルスベクターここで説明した受精卵母細胞の囲卵腔注入は、収集された胚の合計数を基準にして遺伝子胚の 70% 以上が得られた形質転換の胚の生産で起因しました。この結果、以前のレポートに一貫した手順2,7,10、11,12の特異性を例証します。表 1に示したすべてのデータを比較すると、重要な機能が強調表示できます。まず、注入された卵の数は、正常な形態を持っていたまたは文化で数時間後溶解しないすべての挿入された卵に対応しました。注入された卵の 93% は、レンチウイルスベクター囲卵腔内注入による急速な毒性のほぼ完全な欠如を示唆して移植されました。注入された卵の 44% だけ生き残ったし、移植されたので DNA 注入を考慮したとき、状況は劇的に異なるです。さらに、収集した胚移植卵に対する比は、レンチウイルスベクターの悪化の長期毒性がないことを示す 2 つの手順の間と同じです。第二に、注入された卵の数に比べて transgene の統合の胚の数を表現するとき、グローバルの収量は、レンチウイルスベクター注入 DNA 注入と比較して 10 倍以上です。86-fold 違いが同じ Neurog3 増強物の要素を使用して 2 つの手順の間の遺伝子を表現する胚の数を比較するときもあります。
重要なは、遺伝子組み換え生産量は使用されるレンチウイルスベクターの伝達力価の依存するように表示されます。つまり、レンチウイルスベクター制作 10 上記価9 TU/mL はそのような高利回りを得るのに十分な。囲卵腔の噴射量は 10 から 100 までの範囲でプロトコルのセクションで説明した、pl.このボリュームは 10 に 100 アクティブなレンチ ウイルス粒子を表します。レンチウイルスベクターを用いたとき標準的な前核DNA 注射と比較して生まれた動物の同じ量で生成された創設者動物の総数は少なくとも 10 倍高い。さらに、遺伝子発現の浸透度がユビキタスの両方が観察され、CMV プロモーターを除いて特定のプロモーターを細胞にはこのプロトコルで非常に高い。携帯ユビキタス プロモーターへの反対、CMV プロモーター DNA メチル化20は積極的にシャット ダウンと長期式21多能性幹細胞の情報伝達を維持することができないとされています。これは eGFP 発現トランスジェニック胚において細胞の非常に限られた数を説明する可能性があります。したがって、レンチウイルスベクターは transgene の式、セル特定エンハンサーによって制御するトランスジェニック動物の生産にも適応。重要なは、エンハンサー活性体内の画面に、検索地図転写因子結合部位の規定する領域11,12内には、プロトコルを使用できます。この審査方法は、標準的な遺伝子を使用してほとんど実行できます。創設者動物のすべての異なる構成をテストし、統計的有意性に到達するために必要な数の合計は、レンチウイルスベクターを介した遺伝子導入と急速に取得できますに対し注射セッションの数十を必要があります。
標準的な手順とレンチの手法の主な違いの 1 つは、transgene の統合に存在します。前核の注入を使用 transgenes はユニークな軌跡で複数のコピーとしてランダムに統合します。レンチ ウイルスのベクトルを使用して、統合、厳密にランダムなことがなく複数遺伝子 (遺伝子座あたりの 1 つのコピー) で発生します。線形増幅を介した PCR (ラム PCR) を用いた統合サイトを複製することによって + Trono のグループは、遺伝子を受精卵13のクロマチン領域に優先的に統合を示しています。統合バイアスに干渉したり、トランスジェニック マウスの遺伝子発現に寄与しません。レンチ ウイルス伝達 1 つのセル段階の胚の中に統合は、開発中に後で構成のオープン中または大人でないかもしれないクロマチンで発生します。
さらに、第一世代の動物 (F0) や胚に統合された遺伝子のコピー数を分析し、統合された遺伝子の数に大きな変動が観察されます。この研究では、19 の統合されたコピーの平均が発見された Neurog3-ガ-eGFP コンストラクトを使用。この大規模なコピー数は、モザイクの高レベルを反映できます。Sauvainらは、大規模な実行しているレンチウイルスベクターを介したメソッドで生成された F0 動物の統合の軌跡の研究は13をここで説明します。彼らは 70 の個々 の統合サイト 11 F0 動物を追い、F1 後継者に F0 トランスジェニック マウスから各サイトの転送速度を調べた。個々 の統合 transgene の 44% の透過率は、彼らは、いずれか 1 細胞期胚または 2 細胞期で S 期前に S 段階後によく確立された示唆しています。確かに、S 段階の前に統合しながら統合後、S 相はのみ 1 つの娘細胞にそれを伝えるでしょう、両方の娘細胞に統合された遺伝子を送信するでしょう。したがって、個々 の統合 transgenes のモザイクの程度はこの手法を通じて取得したトランスジェニック マウスでは最小です。これはさらに、使用される培養条件で最初胸の谷間の生産の平均時間に対応する最初の 12 h 以内ほとんど統合が発生することを示します。この統合運動が T リンパ細胞22でのレンチの記載と一致です。
重要なは、むき出し統合遺伝子座数が多いとマウス ラインの確立しない妥当でしょう。これらのすべての遺伝子を分離する踏切の数はかなり高いでしょう。これは、遺伝子の影響の急速なスクリーニングのためまたは複数の遺伝子の同時解析のために使用する必要がありますこのメソッドの重要な制限の 1 つを表します。それにもかかわらず、マウス線は、まだ最低統合遺伝子コピー数と F0 動物ものから選択することによって確立できます。
以来前核 DNA 注入方法1の最初の説明は、最初の手順の欠点の多くを回避、改善がなされています。改善の最初のセットは、カセット交換戦略を使用して正確な軌跡で対象の統合に基づいていた。いずれかの CRE を用いて前核注入、統合の DNA 断片と共にフリップまたは PhiC31 リコンビナーゼの両脇に loxP、FRT または attB サイト、それぞれ。このような状況で統合の DNA は同じシュプリンガー特定サイト23,24の両脇に統合された片を使用して交換されます。第一世代動物の 60% までは、このような法を用いたトランスジェニック23を指定できますが、前核注入の技術にリンクされている制限がまだ適用されます。改善の 2 番目のセットは 2 つの環状 DNA を 1 つに統合するフラグメントを運んでいるといずれかメダカ Tol225、1 つのことができます式の細胞質注射基づいて眠れる森の美女26または piggyBac27トランスポサーゼ。これらのメソッドを使用すると、高い利回りが得られる (> 30%)、しかし、もっと重要なは、細胞質内注入が容易に行えるし、レンチ ウイルス ベースのプロトコルとして前核注入による制限を回避します。さらに、細菌人工染色体などの非常に大きな DNA 断片を統合できます。
レンチウイルスベクターを介した遺伝子導入は標準も改善された手順を置き換えられますは明らかです。まだかなり少なくとも遺伝的変異性を持つ動物の適切な数を生成する必要な時間を短縮するために、このメソッドは急速な動物モデルの作製と解析のための強力なツールを表します。さらに、この技術は、任意の遺伝子組み換えの行を含むすべてのマウス系統に直接適用されます。さらに、新しい動物モデルの生成の世界各地の風景がについて CRISPR/Cas9 技術の最近の開発を変更することを言及することが重要です。今日、Cas9 とともにガイド RNA 蛋白質の前核の注射は、ゲノムの生産は 4028の有効性と動物のモデルを編集できます。このアプローチ主、レンチ ウイルス仲介された遺伝子の使用の恩恵を受けることができます。確かに、製品歩留まりも非統合レンチウイルスベクター29一過性両方の Cas9 を表現し、Rna をガイドの使用可能性があります。関連性の高い堅牢な動物モデルを作成する最新の技術の組み合わせは、最も国際的な研究グループに病気の発症と治療法を勉強して恩恵。
開示事項
著者はある利益相反を開示します。
謝辞
レンチ ウイルスのベクトル生産および動物それぞれ住宅での技術支援の原稿と iVector Phenoparc ICM コアの重要な読書、マガリ ・ デュモンとローランド メローニに感謝します。この作品は、Institut Hospitalo ユニヴェルシテール ・ デ ・神経科学 Translationnelles デによって支えられたパリ、Investissements d'Avenir ANR-10-IAIHU-06 IHU-A-ICM。広報協会・ デ ・ ラング ・ フランセーズのための資金を受け取った注ぐ流動デュ Diabète et des 病気 Métaboliques (ALFEDIAM) と共同 JDRF INSERM 付与/。
資料
| Name | Company | Catalog Number | Comments |
| PMSG 50UI | Sigma | G4527 | |
| hCG 5000UI | Sigma | CG5-1VL | |
| NaCl | Sigma | 7982 | |
| 100 mm petri dish | Dutsher | 353003 | |
| 4 wells Nunc dish | Dutsher | 56469 | IVF dish |
| M2 medium | Sigma | M7167 | |
| M16 medium | Sigma | M7292 | |
| 0,22 µm Syringe filter | Dutsher | 146611 | |
| Hyaluronidase Enzyme 30mg | Sigma | H4272 | mouse embryo tested |
| Insulin serynge | VWR | 613-3867 | Terumo Myjector |
| Curved forceps | Moria | 2183 | |
| Curved scissors | Moria | MC26 | |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma | A5177-5EA | |
| Borosilicate glass capillaries | Harvard apparatus | GC 100-10 | |
| Horizontal micropipette puller | Narishige | PN-30 | |
| Microforge | Narishige | MF-900 | |
| Inverted microscope | Nikon | Transferman NK2 5188 | Hoffman modulation contrast illumination is required |
| Micromanipulator | Eppendorf | Celltram air | |
| Controler of holding pipet | Eppendorf | Femtojet | |
| Mineral oil | Sigma | M8410 | mouse embryo tested |
| Microinjector | Eppendorf | Femtojet | Can be used to inject DNA or viral vectors |
| Dumont # 5 forceps | Moria | MC 40 | |
| vannas micro scissors | Moria | 9600 | |
| Isoflurane | centravet | ISO005 | ISO-VET 100% 250ml |
| ocrygel | centravet | OCR002 | |
| Povidone iodure | centravet | VET001 | vetedine 120ml |
| Buprenorphine | centravet | BUP002 | Buprecare 0,3Mg/ml 10ml |
| Tris-HCl | Sigma | T5941 | Trizma hydrochloride |
| EDTA | Sigma | E9884 | |
| SDS | Sigma | 436143 | |
| NaCl | Sigma | S7653 | powder |
| proteinase K | Sigma | P2308 | |
| oneTaq kit | NEB | M0480L | |
| Primers | Eurogentec | ||
| Strip of 8 PCR tube | 4titude | 4ti-0781 | |
| 96 well thermal cycler | Applied Biosystems | 4375786 | Veriti |
| Genomic DNA mini kit | invitrogen | K1820-02 | |
| Nanodrop 2000 | Thermo Scientific | ND-2000C | |
| qPCR Master mix | Roche | 4887352001 | SYBR Green |
| Multiwell plate 384 | Roche | 5217555001 | |
| qPCR instrument 384 well | Roche | 5015243001 | LightCycler 480 |
参考文献
- Gordon, J. W., Scangos, G. A., Plotkin, D. J., Barbosa, J. A., Ruddle, F. H. Genetic transformation of mouse embryos by microinjection of purified DNA. Proceedings of the National Academy of Science USA. 77 (12), 7380-7384 (1980).
- Bock, T. A., Orlic, D., Dunbar, C. E., Broxmeyer, H. E., Bodine, D. M. Improved engraftment of human hematopoietic cells in severe combined immunodeficient (SCID) mice carrying human cytokine transgenes. Journal of Experimental Medicine. 182 (6), 2037-2043 (1995).
- Miyakawa, Y., et al. Establishment of human granulocyte-macrophage colony stimulating factor producing transgenic SCID mice. British Journal of Haematology. 95 (3), 437-442 (1996).
- Hirabayashi, M., et al. A comparative study on the integration of exogenous DNA into mouse, rat, rabbit, and pig genomes. Experimental Animals. 50 (2), 125-131 (2001).
- Isola, L. M., Gordon, J. W. Transgenic animals: a new era in developmental biology and medicine. Biotechnology. 16, 3-20 (1991).
- Zufferey, R., Nagy, D., Mandel, R. J., Naldini, L., Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nature Biotechnology. 15 (9), 871-875 (1997).
- Lois, C., Hong, E. J., Pease, S., Brown, E. J., Baltimore, D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 295 (5556), 868-872 (2002).
- Ewerling, S., et al. Evaluation of laser-assisted lentiviral transgenesis in bovine. Transgenic Research. 15 (4), 447-454 (2006).
- Ritchie, W. A., Neil, C., King, T., Whitelaw, C. B. Transgenic embryos and mice produced from low titre lentiviral vectors. Transgenic Research. 16 (5), 661-664 (2007).
- Park, F. Lentiviral vectors: are they the future of animal transgenesis. Physiological Genomics. 31 (2), 159-173 (2007).
- van Arensbergen, J., et al. A distal intergenic region controls pancreatic endocrine differentiation by acting as a transcriptional enhancer and as a polycomb response element. PLoS One. 12 (2), e0171508 (2017).
- Friedli, M., et al. A systematic enhancer screen using lentivector transgenesis identifies conserved and non-conserved functional elements at the Olig1 and Olig2 locus. PLoS One. 5 (12), e15741 (2010).
- Sauvain, M. O., et al. Genotypic features of lentivirus transgenic mice. Journal of Virology. 82 (14), 7111-7119 (2008).
- Punzon, I., Criado, L. M., Serrano, A., Serrano, F., Bernad, A. Highly efficient lentiviral-mediated human cytokine transgenesis on the NOD/scid background. Blood. 103 (2), 580-582 (2004).
- Zennou, V., et al. The HIV-1 DNA flap stimulates HIV vector-mediated cell transduction in the brain. Nature Biotechnology. 19 (5), 446-450 (2001).
- Miyoshi, H., Blomer, U., Takahashi, M., Gage, F. H., Verma, I. M. Development of a self-inactivating lentivirus vector. Journal of Virology. 72 (10), 8150-8157 (1998).
- Yee, J. K., et al. A general method for the generation of high-titer, pantropic retroviral vectors: highly efficient infection of primary hepatocytes. Proceedings of the National Academy of Science USA. 91 (20), 9564-9568 (1994).
- Castaing, M., et al. Efficient restricted gene expression in beta cells by lentivirus-mediated gene transfer into pancreatic stem/progenitor cells. Diabetologia. 48 (4), 709-719 (2005).
- Gradwohl, G., Dierich, A., LeMeur, M., Guillemot, F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proceedings of the National Academy of Science USA. 97 (4), 1607-1611 (2000).
- Scharfmann, R., Axelrod, J. H., Verma, I. M. Long-term in vivo expression of retrovirus-mediated gene transfer in mouse fibroblast implants. Proceedings of the National Academy of Science USA. 88 (11), 4626-4630 (1991).
- Norrman, K., et al. Quantitative comparison of constitutive promoters in human ES cells. PLoS One. 5 (8), e12413 (2010).
- Vandegraaff, N., Kumar, R., Burrell, C. J., Li, P. Kinetics of human immunodeficiency virus type 1 (HIV) DNA integration in acutely infected cells as determined using a novel assay for detection of integrated HIV DNA. Journal of Virology. 75 (22), 11253-11260 (2001).
- Ohtsuka, M., et al. One-step generation of multiple transgenic mouse lines using an improved Pronuclear Injection-based Targeted Transgenesis (i-PITT). BMC Genomics. 16, 274 (2015).
- Tasic, B., et al. Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proceedings of the National Academy of Science USA. 108 (19), 7902-7907 (2011).
- Sumiyama, K., Kawakami, K., Yagita, K. A simple and highly efficient transgenesis method in mice with the Tol2 transposon system and cytoplasmic microinjection. Genomics. 95 (5), 306-311 (2010).
- Garrels, W., et al. Cytoplasmic injection of murine zygotes with Sleeping Beauty transposon plasmids and minicircles results in the efficient generation of germline transgenic mice. Biotechnology Journal. 11 (1), 178-184 (2016).
- Ding, S., et al. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 122 (3), 473-483 (2005).
- Aida, T., et al. Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biology. 16, (2015).
- Philippe, S., et al. Lentiviral vectors with a defective integrase allow efficient and sustained transgene expression in vitro and in vivo. Proceedings of the National Academy of Science USA. 103 (47), 17684-17689 (2006).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved