
分子軌道 (MO) 理論
概要
ソース: タマラ ・ m ・力、化学のテキサス A & M 大学
このプロトコルは、2 つの金属錯体が配位子 1, 1'-ビス (ジフェニルホスフィノ) フェロセン (dppf) を搭載の合成におけるガイドとして: M (dppf) Cl2M = Ni や Pd。4 座標のこれらの遷移金属の複合体の両方が、彼らは金属センターで異なる形状を示します。分子軌道 (MO) 法を使用すると、 1H NMR とエバンス メソッドと組み合わせて、我々 はこれら 2 つの化合物の幾何学を決定します。
原則
さまざまな化学分子の結合を記述する使用モデルがあります。モデルはシステムの形式であり、したがって強みだけでなく、重要な制限事項があることが重要です。たとえば、ルイス ・ ドット構造、原子が電子を共有する方法を記述するための最も簡単な方法は考慮分子の原子のジオメトリ。原子価殻電子対反発 (VSEPR) 理論は原子のジオメトリを記述する、価電子の同じ数を用いるアイソエレクトロニック種が異なるジオメトリを表わすことができる観察のための説明は行いません。特に遷移金属錯体のこれらのモデルの両方の金属の結合を記述する短い秋します。結晶場理論は、遷移金属錯体に固有接合モデルです。このモデルは、金属センター dまたはf原子軌道の配位子の電場の効果に見えます。これらの原子軌道の縮退の休憩で相互作用の結果。
この動画で MO 理論だけでなくメインのグループ分子の結合を記述するために使用できますが、遷移金属錯体の結合のモデル化にも適している強力なモデルであるに焦点を当てます。ここでは、我々 は金属含有のモーメント図を生成する方法をデモンストレーションします化合物。
MO 理論:
MO 理論では、与えられた化合物中の各原子の原子軌道 (LCAO) の線形結合として化学結合について説明します。LCAOs に起因する MOs は、ジオメトリと (すなわち、方向性と特定の原子によって形成される結束の強さ) 分子の原子の数によって共有電子のエネルギーの両方について説明します。
MO 理論の基礎を確認するには、二原子分子 F2 (図 1の完全図 MO) をまず検討します。フッ素原子は 4 価の原子軌道: 2s、2px、2py、および 2pz。2s軌道はすべて同じエネルギーを持っている、2p原子軌道よりエネルギーで低い。原子軌道の線形結合は、原子軌道のようなエネルギーと対称性に一致すると発生します。この場合、1 個の F 原子の 2s軌道は 2s軌道、他の F 原子と対話します。MO (図 1) を接合 σ の形成にこれらの 2 つの軌道の結果を追加。結合は安定の相互作用と、したがって、結果として得られる σ MO 2s原子軌道のエネルギーを基準にしてエネルギーの下位にあります。2s原子軌道 (図 1) を基準にしてエネルギーの上位である σ * として指定 (不安定) 反結合軌道相互作用の 2s軌道結果を減算します。

図 1.F2の MO の図表。
同様に、2pの原子軌道を接合形と反結合軌道相互作用に結合します。2s軌道のような (これは F F ボンドに沿ってレイアウト)、2pzの原子軌道は σ と σ * の相互作用を形成します。2pxと 2py原子軌道を考える場合、我々 は彼らが異なったタイプの接合および反結合軌道を形成参照してください相互作用、π、π * をそれぞれと呼ばれます (図 1)。区別するために σ および π 結合 σ 結合性軌道は流行り廃りの軸に円筒対称な π 軌道は流行り廃りの軸方向の節面を持っているので簡単です。Σ 結合を形成する原子軌道間の空間的重なりが π 結合を形成する原子軌道間の空間的重なりより大きいです。したがって、結果 π、π * MOs が少ない安定し不安定になり、それぞれ、2pz原子軌道によって形成される σ と σ * の MOs と比較しています。我々 は、2 個の F 原子の価電子を持つ MOs をし入力できます。
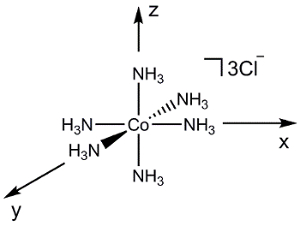
[Co (NH3)6] Cl3 (図 2) などのより複雑な分子を考えます。(一度に 2 原子間原子軌道の重なりを考慮して) 上記と同じプロセスを使用するなら、この分子の MO 図の生成は非常に難しいでしょう。代わりに、最初に、配位子の対称性適応線形結合 (SALC) を生成するのにグループ理論を使用できます。我々 は結合/反 bonding の相互作用金属の原子軌道と結果の SALCs の間にフォームを決定するのに対称性を使用してできます。
| Oh | E | 8C3 | 6C2 | 6C4 | 3C2' | 私 | 6S4 | 8秒6 | 3 σh | 3 σd | ||
| 1 g | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | x2+ y2+ z2 | |
| 2 g | 1 | 1 | -1 | -1 | 1 | 1 | -1 | 1 | 1 | -1 | ||
| Eg | 2 | -1 | 0 | 0 | 2 | 2 | 0 | -1 | 2 | 0 | (2z2x2-y2、2y2x) | |
| T1 g | 3 | 0 | -1 | 1 | -1 | 3 | 1 | 0 | -1 | -1 | (RxRyRz) | |
| T2 g | 3 | 0 | 1 | -1 | -1 | 3 | -1 | 0 | -1 | 1 | (xy、yz xz) | |
| 1 u | 1 | 1 | 1 | 1 | 1 | -1 | -1 | -1 | -1 | -1 | ||
| 2 u | 1 | 1 | -1 | -1 | 1 | -1 | 1 | -1 | -1 | 1 | ||
| Eu | 2 | -1 | 0 | 0 | 2 | -2 | 0 | 1 | -2 | 0 | ||
| T1 u | 3 | 0 | -1 | 1 | -1 | -3 | -1 | 0 | 1 | 1 | (x、y、z) | |
| T2 u | 3 | 0 | 1 | -1 | -1 | -3 | 1 | 0 | 1 | -1 | ||
| Γ赤 | 6 | 0 | 0 | 2 | 2 | 0 | 0 | 0 | 4 | 4 |
Γ赤 =A1 g+ Eg+ T1 u
図 2 。[Co (NH3)6] Cl3のリガンド原子軌道の線形結合。
[Co (NH3)6]3 +SALCs を生成するため我々 は無機化学シリーズのビデオ「集団理論」で説明されている同様手順に従います。
1 分子の点グループを決定します。
2 配位子の原子軌道の簡約表現を生成します。
3. 既約表現に還元できない表現を減らします。
[Co (NH3)6]3 +ポイント グループOhです。金属センターで接合について考えているだけ、ので我々 は単にそれぞれの NH3配位子の 2s軌道を検討できます。私たちの手順 1-3 N 2s軌道関数の簡約表現は Γ赤の場合 =1 g +g E T1 u (図 2)。A1 gセットを表す 1 SALC、間 Egと T1 uセット実際にそれぞれを表します 2 と 3 の SALCs、6 SALCs (陽イオン [Co (NH3)6]3 +の配位子の同じ数) の合計を与えます。Egセットで 2 SALCs は同じ対称性があるし、(同じ T1 uセット 3 SALCs について言うことが) Co の原子軌道とやり取りするとき縮退 MOs になります。文字表を使用すると、図 2に、Co の原子軌道がOhポイント グループに変換方法を判断できます。たとえば、z2 d と dx2– y2軌道、Egセットを形成します。2 の配位子 SALCs Eg対称性を持つので、それらの SALCs は dz2 dx2 –y2 Co 原子軌道と結合/反 bonding 相互作用を形成します。図 3に示す遷移金属錯体の MO 図を生成すべての Co の原子価原子軌道の同じ方法で継続し、私たち。気付いた残りd-軌道 (dxy、yzd は、dxz) セット (T2 g) として変換はありません適切な対称性は、SALC を一致します。これらの原子の軌道「非結合」MOs になります。言い換えれば、彼らはこの遷移金属錯体における配位子の結合に参加しません。
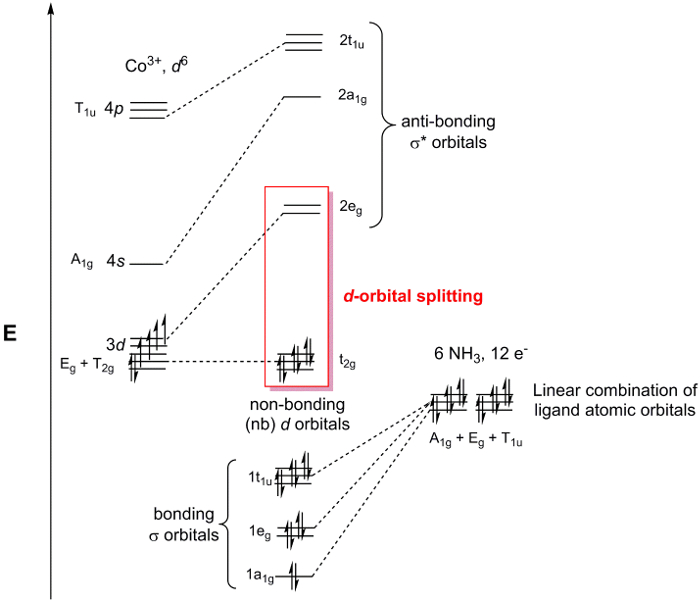
図 3.[Co (NH3)6] Cl3MO 図。
非結合のdは、図 3に強調表示されます-軌道とdσ * 軌道-軌道の文字。Dと呼ばれますときにモーメント図を全体から分離して考えられるこの MOs のグループ-軌道遷移金属の図の複雑な分割します。D以来の軌道図を分割を含む化学や錯体のスペクトルを理解する最も重要な軌道は、通常、ヒトと、LUMO 化学者は多くの場合dを参照してください-軌道ダイアグラム全体を MO ではなくダイアグラムを分割します。便利なd-金属センター de-の数で軌道図を分割できる満ちるリガンド ベースの電子は常に MO の図 σ ベース MOs を埋めるので。
を考慮した、d-軌道分割図 M(dppf) Cl2:
単純な 4 座標金属複雑な MX4を検討してください。MX4することができます 2 つのジオメトリの存在: 四面体または正方形平面。D-軌道分割図のポイント グループTd (四面体) とD4 h (平面) は、図 4に示します。我々 はまだこれらのdを使用することができます M (dppf) Cl2一般的な金属錯体が 4 と同じ配位子はありません、したがってポイント グループTdやD4 hではない-軌道、として図を分割モデル d-を記述する 2 つの使用可能なジオメトリの軌道 MOs。
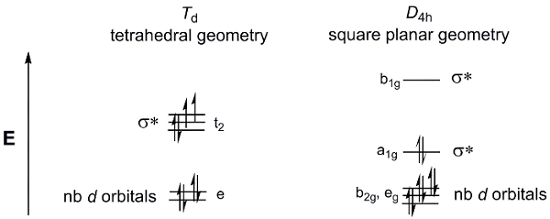
図 4 。Dの軌道ポイントの図を分割グループTd (四面体) とD4 h (平面)。
Dを考えます-M (dppf) Cl2の電子カウント。Ni や Pd は、周期的なテーブルのグループ 10。したがって、両方が同じ酸化状態 (2 +) とd-電子カウント (d8)。我々 は 2 つのdを入力する場合-8 電子上の軌道分割図、四面体のモーメント図は常磁性種と一貫性のある正方形平面のジオメトリが反磁性複合体で結果がわかります。どのジオメトリは精力的に支持されるを決定するにいくつかの要因があります。平面幾何学の正方形平面の幾何学はもっと電子的に好まれていることを示す反結合性軌道に少数の電子があります。しかし、我々 はまたペア電子に必要なエネルギーを考慮する必要があります。正方形平面の分子にエネルギーをペアリング電子は少ない完全に満たされた軌道を持つ正四面体型分子のより高いです。最後に、我々 はσ * d量を考慮する必要-軌道が不安定になります。大きい金属原子配位子、高いエネルギー σ * dの結果を大きな空間の重複している-軌道。
最後に、我々 はまた立体反発力からエネルギーの貢献を考慮する必要があります。四面体の幾何学はより多く正方形平面ジオメトリ (90 °) と比較して立体(109.5 ° の角度) に好まれています。したがって、どのジオメトリは M (dppf) Cl2M のアイデンティティを考えるより有利に影響を与えるいくつかの対立要因があります。
NMR を使用してこれらの 2 つのジオメトリを区別出来る。診断1H NMR を遵守します分子が平面の場合、反磁性種。分子が四面体の場合は、 1H NMR 信号を常磁性が遵守します。エバンス メソッドを使用して、最後に、(詳細については、「エバンス法」ビデオを見なさい) 常磁性種のソリューション磁気モーメントを決定しますします。
手順
注: 安全のため Schlenk ラインの安全は実験を行う前に審査する必要があります。ガラス製品は、使用する前に星のき裂検査必要があります。リキッド N2を使用している場合に、Schlenk ライン トラップに O2がない凝縮されて確保するため注意が必要があります。温液体 N2 O2凝縮し、有機溶媒存在下で爆発。O2が凝縮されているまたは青色の液体はコールド トラップで観察されることが疑われる場合は動的真空下ではコールド トラップを残します。リキッド N2トラップを取除くか、または真空ポンプ.をオフにします。ポンプに蒸発すれば液体 O2時間をかけてO2のすべてが蒸発した後、液体 N2トラップを削除しても安全はありません。詳細については、「Ti(III) メタロセン使用シュレンク管技術の合成」のビデオを参照してください。1
1. ni 合成 (dppf) Cl2シュレンク管と Pd (dppf) Cl2のセットアップ
注: より詳細な手順を参照してください有機化学の必需品シリーズの「Schlenk ライン転送の溶媒」ビデオ).
- 圧力解放バルブを閉じます。
- N2ガス、真空ポンプをオンにします。
- Schlenk としてラインの真空に達するその最低気圧、液体 N2またはドライアイス/アセトンでコールド トラップを準備します。
- コールド トラップを組み立てます。
2. Ni (dppf) Cl2 (図 5) 不活性/嫌気条件下での合成
注中 Ni (dppf) Cl2の合成は、好気性条件下で行うことが、嫌気的条件で行ったときに高い利回りが得られます。
- 3 首フラスコに 550 mg dppf (1 モル) とイソプロパノールの 40 ミリリットルを追加します。
Dppf シグマ アルドリッチから購入することができますまたは文献の方法を使用して合成されるに注意してください。2 - コンデンサーと真空アダプター 3 首フラスコのセンター ネックを合わせてください。1 ガラス栓、ゴムキャップ 1 2 つの残りの首に合います。
- 「ベント」コンデンサーの上部に真空アダプターを 15 分使用溶剤を N2ガスをバブリングによってソリューションをドガ
- N2シュレンク管を使用するコンデンサーの上部に真空アダプターを接続します。
- 90 ° C に設定水浴の三首フラスコを加熱開始
- 丸底フラスコ一つの首で 237 mg NiCl2·6H2O を追加(1 モル) を 4 mL のイソプロパノール (試薬グレード) とメタノール (試薬等級) の 2:1 の混合物の。まで (約 1 分) を溶解している Ni 塩のすべての得られた混合物の超音波照射します。
注: 超音波発生装置が利用できない場合は優しく水浴中で混合物を加熱します。 - 5 分の混合物を通して N2ガスをバブリングによって Ni ソリューションをドガします。
- カニューレ振込三首丸底フラスコに NiCl2·6H2O ソリューションを追加します。
- 90 ° C で 2 時間還流反応を可能します。
- 氷浴で冷却し反応を許可します。ガラスフリット漏斗を通して真空ろ過によって結果緑沈殿物を分離します。
- ヘキサンの 10 mL に続いて、冷たいイソプロパノール 10 mL で製品を洗います。
- NMR サンプルを準備する前に製品の空気を乾燥を許可します。
- クロロホルム -d 1H NMR 製品を取る。
- 1H NMR が常磁性種の指標の場合は、エバンス メソッド、手順 4 の指示に従って、NMR を準備します。

図 5。Ni (dppf) Cl2の合成。
3. Pd (dppf) Cl2 (図 6)1の合成
注: Pd (dppf) Cl2の合成のための標準 Schlenk ライン技術の使用 (「Ti(III) メタロセン使用シュレンク管技術の合成」のビデオを参照してください).
メモしながら Pd (dppf) Cl2の合成は、好気性条件下で行うことが、嫌気的条件で行ったときに高い利回りが得られます。
- Schlenk フラスコに 550 mg (1 モル) dppf と 383 mg (1 モル) bis(benzonitrile)palladium(II) 塩化物を追加し、溶媒のカニューレ転送 Schlenk フラスコを準備します。
- カニューレ振込 Schlenk フラスコに脱トルエン 20 mL を追加します。
- 少なくとも 12 時間室温で撹拌する反応を許可します。
- ガラスフリット漏斗を通して真空ろ過によって結果のオレンジ色沈殿物を分離します。
- 続いてヘキサン (10 mL) トルエン (10 mL) を持つ製品を洗います。
- NMR サンプルを準備する前に製品の空気を乾燥を許可します。
- クロロホルム -d 1H NMR 製品を取る。
- 1H NMR が常磁性種の指標の場合は、手順 4 に記載されている指示に従ってエバンス メソッドの NMR を準備します。

図 6。Pd (dppf) Cl2の合成。
4. エバンス メソッドのサンプルの準備
注: より詳細な手順、「エバンス法」のビデオを参照してください。
- シンチレーション バイアルでクロロホルム-dの 50: 1 (ボリューム: ボリューム) ソリューションの準備: trifluorotoluene。ピペット 2 mL、重水素化溶媒とこれには、trifluorotoluene の 40 μ L を追加します。バイアルをキャップします。
注: この例では、我々 使用する19F-NMR 常磁性種の存在下で trifluorotoluene で F 信号の変化を観察します。 - このソリューションにより、キャピラリーの挿入を準備します。
- 新しいシンチレーション バイアルに 10-15 mg の常磁性のサンプルの重さし、質量に注意してください。
- ピペット ~ 600 μ L 常磁性種を含むバイアルに準備された溶媒の混合物の。質量に注意してください。ソリッドが完全に分解することを確認します。
- 標準的な NMR チューブで慎重に壊れないように、角度で毛細管挿入をドロップします。
- NMR 管に常磁性種を含む溶液をピペットします。
- 取得し、標準19F NMR スペクトルを保存します。
- プローブの温度に注意してください。
- 高周波に注意してください。
結果
Pd (dppf) Cl2:
1H NMR (クロロホルム-d、400 MHz、δ、ppm): 7.44、7.89, 4.42 (ベータ-H) 4.22 (アルファ-H) 7.54 (芳香族)3。
Ni (dppf) Cl2:
1H NMR (クロロホルムは δ をd、300 MHz、ppm): 20.85、10.04、4.23、3.98、1.52、-3.31、-7.10。
エバンス法、trifluorotoluene の19F シフトを見て:
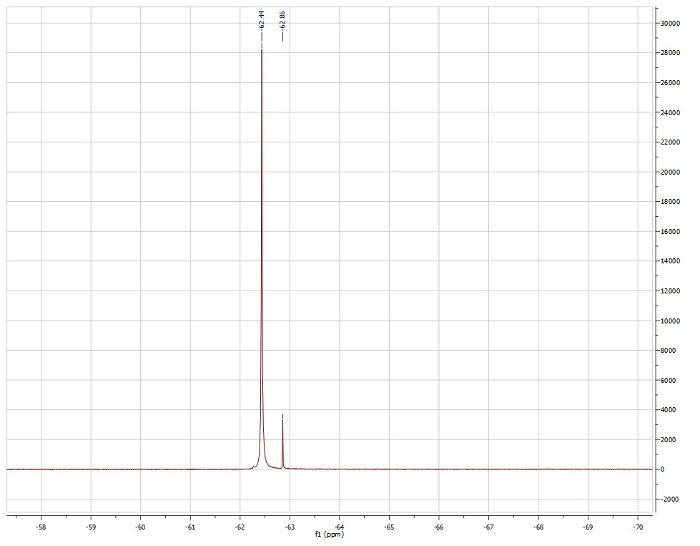
観察μeff = 3.15 μb
サンプルの質量: 9.5 mg
ソリューション (クロロホルムd + trifluorotoluene) の固まり: 0.8365 g
プローブの温度: 296.3 K
NMR フィールド (MHz): 470.06
報告 μeff = 3.39 μb。4
S = 1 (予測に基づいて四面体幾何学、図 4)、理論的な μeff = 2.83 μb。
S = 3/2、理論的な μeff = 3.46 μb。
1H NMR データに基づいて、我々 は Pd (dppf) Cl2は反磁性とそのため正方形平面ジオメトリを示すことを参照してください。1H NMR の Ni (dppf) Cl2は常磁性は、Ni センターで四面体です。エヴァンのメソッドは、Ni (dppf) Cl2は常磁性、3.15 μbに近いソリューション磁気モーメントを示す文献報告この化合物の値を確認します。Ni が小さいので、sterics は、Ni (dppf) Cl2四面体を作って、正方形平面のジオメトリに関連付けられている任意の電子安定化を上回る。一方、Pd が大きく、したがってより高いエネルギー σ * dは、-軌道。この場合、電子安定化は、Pd (dppf) Cl2で Pd で平面幾何学の結果として、立体反発力を大きく上回る。
申請書と概要
このビデオでは、遷移金属錯体における結合のモデルとしての MO 理論の使用方法を説明しました。一般式 M (dppf) Cl22 錯体を合成しました。場合 M = Ni 4 座標複雑な展示四面体の幾何学。Ni 原子を大きく遷移金属 (Pd) に置き換えて、分子は平面ジオメトリを取ります。
以前は、有機金属化学の分野で重要な役割フェロセンについて学びました。置換フェロセン、dppf を含む、キレート配位子の 1st2nd、3rd行遷移金属として使用されます。結果として得られる錯体は均一系触媒で使用される (すなわち。、[1,1'-bis(diphenylphosphino)ferrocene]palladium(II) 二塩化、Pd (dppf) Cl2、C C および C-ヘテロ原子結合形成反応の触媒である)。
遷移金属錯体の結合を理解の構造と反応性を説明するために重要です。MO 理論の強みの 1 つは、遷移金属錯体の反応性を説明するために使用できる良いモデルを提供することです。多くの場合、金属のセンター、分子によって示される任意の反応性の場所です。したがって、それは図に要約d-軌道分裂分子軌道理論 (図 3) から派生した金属センターで電子密度の画像を得ることが大切です。Dで MOs を行うだけではなくお知らせ-軌道の分割図展示主d-軌道文字 (σ * 軌道が原子のdのエネルギーに最も近い-金属、従ってそれらの MOs の電子密度のほとんどの軌道は金属原子を中心として)、ヒトと分子の LUMO も分割図が含まれています。したがって、発生する化学は直接dに影響-軌道分子の図を分割します。
参考文献
- Corain, B., Longato, B., Favero, G. Heteropolymetallic Complexes of 1,1’-Bis(diphenylphosphino)ferrocene (dppf). III*. Comparative Physicochemical Properties of (dppf)MCl2 (M = Co, Ni, Pd, Pt, Zn, Cd, Hg). Inorg Chim Acta. 157, 259-266 (1989).
- Cullen, W. R., Einstein, F. W. B., Jones, T., Kim, T.-J. Structures of three hydrogenation catalysts [(P-P)Rh(NBD)]ClO4 and some comparative rate studies where (P-P) = (η5-R1R2PC5H4)(η5-R3R4PC5H4)Fe (R1 = R2 = R3 = R4 = Ph, R1 = R2 = Ph, R3 = R4 = CMe3, R1 = R3 = Ph, R2 = R4 = CMe3). Organometallics. 4(2), 346-351 (1983).
- Colacot, T. J., C.-Olivares, R., H.-Ortega, S. Synthesis, X-ray, spectroscopic and a preliminary Suzuki coupling screening studies of a complete series of dppfMX2 (M = Pt, Pd, X = Cl, Br, I). J Organomet Chem. 637-639, 691-697 (2001).
- Rudie, A. W., Lichtenberg, D. W., Katcher, M. L., Davison, A. Comparative Study of 1,1’-bis(diphenylphosphino)cobaltocinium hexafluorophosphate and 1,1’-bis(dipenylphosphino)ferrocene as Bidentate Ligands. Inorg Chem. 17(10), 2859-2863, 1978.
タグ
スキップ先...
このコレクションのビデオ:

Now Playing
分子軌道 (MO) 理論
Inorganic Chemistry
35.4K 閲覧数

シュレンク管を用いた Ti(III) メタロセンの合成
Inorganic Chemistry
31.6K 閲覧数

グローブ ボックスと不純物センサー
Inorganic Chemistry
18.6K 閲覧数

昇華によってフェロセンの精製
Inorganic Chemistry
54.7K 閲覧数

エバンス メソッド
Inorganic Chemistry
68.6K 閲覧数

単結晶および粉末 x 線回折
Inorganic Chemistry
104.7K 閲覧数

電子常磁性共鳴 (EPR) 分光法
Inorganic Chemistry
25.5K 閲覧数

メスバウアー分光法
Inorganic Chemistry
22.0K 閲覧数

Ph3P BH3ルイス酸-塩基相互作用
Inorganic Chemistry
38.9K 閲覧数

フェロセンの構造
Inorganic Chemistry
79.6K 閲覧数

群論の赤外分光法への応用
Inorganic Chemistry
45.5K 閲覧数

Quadruply 金属-金属接合外輪
Inorganic Chemistry
15.3K 閲覧数

色素増感太陽電池
Inorganic Chemistry
15.8K 閲覧数

酸素運ぶのコバルト (ii) 錯体の合成
Inorganic Chemistry
51.7K 閲覧数

根本的な重合反応の光化学開始
Inorganic Chemistry
16.8K 閲覧数
Copyright © 2023 MyJoVE Corporation. All rights reserved