
Standard interni
Panoramica
Fonte: Laboratorio del Dr.B. Jill Venton - Università della Virginia
L'obiettivo di molte analisi chimiche è un'analisi quantitativa, in cui viene determinata la quantità di una sostanza in un campione. Per calcolare con precisione la concentrazione di uno sconosciuto da un campione, è fondamentale un'attenta preparazione del campione. Ogni volta che un campione viene maneggiato o trasferito, parte del campione può andare perso. Esistono tuttavia strategie per ridurre al minimo la perdita di campioni. Esistono anche strategie per far fronte alla perdita del campione e fare ancora misurazioni accurate della concentrazione.
Per ridurre al minimo la perdita di campioni, l'ideale è ridurre al minimo il numero di passaggi di gestione e trasferimento del campione. Ad esempio, la massa di un campione solido direttamente in un pallone in cui verrà realizzata una soluzione riduce una fase di trasferimento. Se è necessario trasferire da un pallone all'altro e si sta facendo una diluizione, il triplo risciacquo della vetreria aiuta a garantire che tutto il campione venga trasferito. Altre strategie sono più specifiche per il campione. Ad esempio, i campioni che assorbono al vetro, come le proteine, potrebbero essere gestiti meglio in tubi monouso in polipropilene. I tubi non sono idrofili, quindi se una piccola quantità di campione deve essere pipettata in acqua, è meglio aver già aggiunto l'acqua al tubo, in modo che il campione possa essere pipettato direttamente nel solvente. Potrebbe essere meglio concentrarsi, piuttosto che asciugare completamente un campione, a causa delle perdite da insolubilità dopo la reidratazione.
Un'altra fonte di perdita del campione è attraverso manipolazioni incomplete del campione. Ad esempio, se viene utilizzata una procedura di derivatizzazione e la derivatizzazione è incompleta, l'intera quantità di campione non viene osservata. Errori come questo sono errori sistematici e possono essere risolti correggendo il problema, ad esempio modificando la procedura di derivatizzazione. Un'altra causa di errore sistematico nelle misurazioni sono gli effetti della matrice. Questi possono interferire con la misurazione di determinate sostanze e l'esecuzione di calibrazioni nella stessa matrice del campione può ridurre questo effetto.
L'analisi quantitativa viene in genere eseguita utilizzando standard esterni o interni. Per gli standard esterni, viene effettuata una curva di calibrazione misurando diverse concentrazioni note dell'analita di interesse. Quindi, l'esempio viene eseguito separatamente dallo standard. Per gli standard interni, lo standard si trova nello stesso campione dell'analita di interesse, consentendo di eseguire la misurazione contemporaneamente. Tipicamente, viene aggiunta una specie diversa chiamata standard interno e viene calcolato il rapporto tra la risposta per quello standard interno e l'analita. L'idea è che il rapporto della risposta, chiamato fattore di risposta, sia proporzionale alla loro concentrazione. Mentre il metodo deve essere in grado di distinguere tra l'analita di interesse e lo standard interno, eventuali perdite di campione che si verificano dopo l'aggiunta dello standard interno dovrebbero essere simili per entrambe le sostanze e quindi il rapporto della risposta rimane lo stesso. Un caso speciale di utilizzo di standard interni è il metodo delle aggiunte standard, in cui quantità crescenti di analita vengono aggiunte alla soluzione e la quantità originale di analita viene retro-calcolata. Gli standard interni possono essere utilizzati in cromatografia, elettrochimica e spettroscopia.
Principi
Uno standard interno è una sostanza aggiunta in quantità costante a un campione, in bianco e standard in un'analisi. Uno standard interno può compensare errori sia sistematici che casuali. Ad esempio, se ci sono fluttuazioni dello strumento che causano errori casuali nella misurazione, queste fluttuazioni dovrebbero essere le stesse sia per lo standard interno che per l'analita e quindi il rapporto tra i segnali non cambia. Per errori sistematici, come gli effetti matriciali del solvente, finché l'effetto è uguale sia per lo standard che per l'analita, il rapporto è di nuovo inalterato.
Lo svantaggio degli standard interni è che è difficile trovare uno standard interno adatto. Lo standard interno deve avere un segnale simile all'analita, ma abbastanza diverso da poter essere distinto dallo strumento. Inoltre, lo standard interno non dovrebbe essere presente nella matrice del campione, in modo che lo standard aggiunto sia l'unica fonte dello standard. Occasionalmente, un costituente principale di un campione che è costante in concentrazione può essere usato come standard interno invece di uno standard aggiunto, ma la concentrazione deve essere ben nota per quel costituente. Lo standard interno non dovrebbe inoltre sopprimere o migliorare il segnale dell'analita.
Nella cromatografia, una delle maggiori fonti di errore è spesso l'iniezione. Se vengono utilizzate iniezioni manuali, possono esserci errori nel caricare correttamente la siringa. I volumi sono tipicamente piccoli (~ 1 μL), quindi ci sono incertezze nell'iniettare in modo riproducibile questo piccolo volume, spesso un paio di percentuali di deviazione standard relativa (RSD). Nella gascromatografia (GC), il campione viene iniettato in una porta riscaldata e l'evaporazione dalla punta dell'ago può provocare variazioni di volume iniettato. I campionatori automatici aiutano sia con l'errore nel caricamento della siringa che con l'errore nell'iniezione rapida per evitare l'evaporazione in GC, ma l'errore può ancora essere dell'1-2% RSD. Con la cromatografia, l'area del picco viene generalmente utilizzata al posto dell'altezza del picco, poiché i picchi diventano più ampi e più corti con il tempo, ma l'area del picco è costante. Pertanto, per gli standard interni, i rapporti delle aree di picco vengono utilizzati nella cromatografia anziché nelle altezze dei picchi.
Per una calibrazione con uno standard interno, viene calcolato un fattore di risposta. Il fattore di risposta (R) è il rapporto dei picchi rispetto al rapporto delle concentrazioni dove X è l'analita e IS è lo standard interno.

Per la cromatografia viene utilizzata l'area (A). Il fattore di risposta può essere calcolato da un grafico di calibrazione di AX/ AIS vs CX/ CIS, dove il fattore di risposta è la pendenza e l'intercetta y è assunta come 0. Una volta che il fattore di risposta è noto per gli standard, la risposta dell'ignoto può essere calcolata dal rapporto di area misurato dall'esperimento.
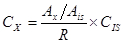
Procedura
1. Corretta gestione del campione: creazione di una soluzione
- Prendi un becher pulito e ammassa la quantità corretta di campione in esso. Registrare la massa effettiva utilizzata. In questo esempio, una soluzione di adenina viene realizzata in un pallone volumetrico per l'uso come standard interno per l'analisi successiva. La massa di adenina è di 100 mg. Non ammassare direttamente in un matraccio volumetrico perché ha un collo lungo e l'adenina non può essere facilmente aggiunta o rimossa.
- Aggiungere circa 25 ml di solvente (in questo caso dimetilsolfossido (DMSO)) al becher e lasciarlo mescolare per sciogliersi. In questo esempio, la soluzione finale è fatta in un matraccio volumetrico da 50 ml, quindi aggiungi solo circa 25 ml in modo che il becher possa essere risciacquato e la soluzione fatta fino al volume finale.
- Una volta che il solido si è sciolto, versare la soluzione nel matraccio volumetrico.
- Risciacquare il becher e la barra di agitazione con piccole quantità di solvente, circa 10 ml, e versare il risciacquo nel matraccio volumetrico. Ripeti altre due volte. Ciò consente di garantire il corretto trasferimento della soluzione.
2. Preparazione di una curva di calibrazione standard interna
- Preparare i campioni standard desiderati per l'analisi gascromatografica. In questo esempio, la caffeina viene estratta dal caffè usando acetonitrile e quindi l'adenina viene utilizzata come standard interno per la misurazione.
- Per i campioni di caffeina, pesare la quantità di campione necessaria per fare 1 mg / mL di campione. Se si utilizza un matraccio volumetrico da 10 ml, ovvero 10 mg.
- Pesare l'analita in un becher, aggiungere qualche mL di solvente (qui metanolo) per sciogliere, quindi trasferire quantitativamente al matraccio volumetrico usando 3 risciacqui.
- Fai altri 3 standard in modo simile con 0,2, 0,5 e 2 mg / ml di caffeina.
- Mettere 1 mL di ogni standard di caffeina in un flaconcino campione.
- Aggiungere 0,2 mL di 2 mg/mL di standard interno di adenina a ciascun flaconcino campione.
- Esegui l'esperimento di gascromatografia con ogni standard di caffeina. Per ogni cromatogramma, calcolare il rapporto tra le aree di picco per la caffeina rispetto allo standard.
- Crea un grafico del rapporto area rispetto al rapporto di concentrazione. La pendenza di quel grafico è il fattore di risposta.
3. Preparazione di un campione reale con standard interno per gascromatografia
- Preparare il campione desiderato per l'analisi gascromatografica. In questo esempio, la caffeina viene estratta dal caffè usando l'acetrontrile.
- Per il campione di caffè, ammassare 2 g di caffè in un becher da 100 ml. Registrare il peso esatto del caffè.
- Aggiungere 20 ml di acetonitrile al becher.
- Lasciare riposare per 20 minuti, mescolando frequentemente.
- Filtrare i fondi di caffè usando una carta da filtro in un imbuto.
- Risciacquare la carta da filtro 3x con piccole quantità di acetonitrile (5 ml).
- Misurare il volume finale del filtrato. Dovrebbe essere di circa 35 ml.
4. Eseguire il campione e calcolare la concentrazione
- Prendere 1 mL del campione di estratto di caffè e aggiungere 0,2 mL dello standard interno in un flaconcino. Posizionare il flaconcino nel rack del campionatore automatico.
- Eseguire un'analisi GC del campione. Assicurarsi che le condizioni GC siano tali che la caffeina e l'adenina si sepatrino. In questo esempio, le separazioni isotermiche vengono eseguite a 200 °C.
- Dopo l'analisi, calcolare l'area di picco sia per il picco standard interno che per il picco analita. Usando i loro rapporti, calcola la quantità di caffeina nel campione.
5. Risultati: Analisi GC della caffeina con standard interno
- L'analisi GC della caffeina è mostrata nella Figura 1. L'adenina è usata come standard interno. Il rapporto tra le aree di picco può essere misurato e tracciato rispetto al rapporto tra le concentrazioni. La pendenza del grafico è il fattore di risposta (in questo caso 1.8).
- La Figura 2 mostra un cromatogramma di un campione di caffè con standard interno di adenina. Il rapporto tra l'area di picco è 1,78. Utilizzando il fattore di risposta e la concentrazione nota di adenina (0,33 mg/mL), la concentrazione di caffeina nel campione sconosciuto è calcolata in 0,33 mg/mL.

Figura 1. Grafico di calibrazione utilizzando uno standard interno. Un grafico dei rapporti di area vs rapporti di concentrazione per 3 campioni standard di caffeina (1, 0,5 e 0,2 mg / mL) con 0,33 mg / mL di standard interno di adenina aggiunti a ciascuno. La pendenza della linea è 1,8, che è il fattore di risposta.
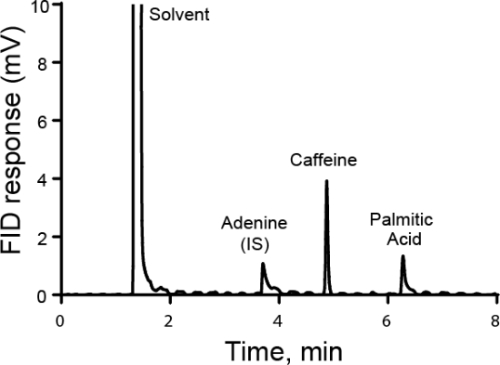
Figura 2. Cromatogramma del caffè con standard interno di adenina. Un grafico della risposta del rilevatore FID ai campioni. I tre picchi principali sono l'adenina (IS), la caffeina e l'acido palmitico.
Applicazione e Riepilogo
Gli standard interni sono utilizzati in molti campi, tra cui la spettroscopia e la cromatografia. In spettroscopia, gli standard interni possono aiutare a correggere errori casuali dovuti a cambiamenti nell'intensità della sorgente luminosa. Se una lampada o un'altra sorgente luminosa ha una potenza variabile, influenzerà l'assorbimento e, di conseguenza, l'emissione di un campione. Tuttavia, il rapporto tra uno standard interno e l'analita rimarrà costante, anche se la sorgente luminosa non lo fa. Un esempio di questo è l'utilizzo del litio (Li) come standard interno per l'analisi del sodio in un campione di sangue mediante spettroscopia di fiamma. Li è chimicamente simile al sodio, ma non si trova nativamente nel sangue.
Per la cromatografia, gli standard interni sono spesso utilizzati sia nella gascromatografia che nella cromatografia liquida. Per le applicazioni con spettrometria di massa come rivelatore, lo standard interno può essere un analita etichettato isotopicamente, in modo che il peso molecolare (MW) sia diverso dall'analita di interesse. Gli standard interni sono comunemente usati nelle analisi farmaceutiche o ambientali.
Tags
Vai a...
Video da questa raccolta:

Now Playing
Standard interni
Analytical Chemistry
204.6K Visualizzazioni

Preparazione del campione per la caratterizzazione analitica
Analytical Chemistry
84.6K Visualizzazioni

Metodo delle aggiunte standard
Analytical Chemistry
319.8K Visualizzazioni

Curve di calibrazione
Analytical Chemistry
796.1K Visualizzazioni

Spettroscopia ultravioletta/visibile (UV-VIs)
Analytical Chemistry
622.9K Visualizzazioni

Spettroscopia Raman per analisi chimiche
Analytical Chemistry
51.2K Visualizzazioni

Fluorescenza a raggi X (XRF)
Analytical Chemistry
25.4K Visualizzazioni

Gascromatografia con rivelatore a ionizzazione di fiamma
Analytical Chemistry
281.8K Visualizzazioni

Cromatografia liquida ad alta prestazione (HPLC)
Analytical Chemistry
384.1K Visualizzazioni

Cromatografia a scambio ionico
Analytical Chemistry
264.3K Visualizzazioni

Elettroforesi capillare
Analytical Chemistry
93.7K Visualizzazioni

Introduzione alla spettrometria di massa
Analytical Chemistry
112.2K Visualizzazioni

Microscopia elettronica a scansione (SEM)
Analytical Chemistry
87.1K Visualizzazioni

Misurazioni elettrochimiche di catalizzatori supportati mediante l'utilizzo di un potenziometro/galvanometro
Analytical Chemistry
51.3K Visualizzazioni

Voltammetria ciclica
Analytical Chemistry
124.9K Visualizzazioni
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati