Microscopia confocal de fluorescência: uma técnica para determinar a localização de proteínas em fibroblastos de camundongos
Visão Geral
Fonte: Dominique R. Bollino1, Eric A. Legenzov2, Tonya J. Webb1
1 Departamento de Microbiologia e Imunologia, Faculdade de Medicina da Universidade de Maryland e o Centro de Câncer Integral Marlene e Stewart Greenebaum, Baltimore, Maryland 21201
2 Center for Biomedical Engineering and Technology, University of Maryland School of Medicine, Baltimore, Maryland 21201
A microscopia de fluorescência confocal é uma técnica de imagem que permite maior resolução óptica em comparação com a microscopia convencional de epifluorescência "campo largo". Microscópios confocal são capazes de obter uma resolução óptica x-y melhorada através da "varredura a laser", tipicamente um conjunto de espelhos controlados por tensão (espelhos galvanômetros ou "galvo") que direcionam a iluminação a laser para cada pixel da amostra de cada vez. Mais importante, os microscópios confocal atingem uma resolução z-axial superior usando um pinhole para remover a luz de foco originária de locais que não estão no plano z que está sendo escaneado, permitindo assim que o detector colete dados de um z-plane especificado. Devido à alta resolução z alcançável na microscopia confocal, é possível coletar imagens de uma série de z-planes (também chamado de z-stack) e construir uma imagem 3D através de software.
Antes de discutir o mecanismo de um microscópio confocal, é importante considerar como uma amostra interage com a luz. A luz é composta de fótons, pacotes de energia eletromagnética. Um fóton que implica em uma amostra biológica pode interagir com as moléculas que compõem a amostra de uma das quatro maneiras: 1) o fóton não interage e passa pela amostra; 2) o fóton é refletido/espalhado; 3) o fóton é absorvido por uma molécula e a energia absorvida é liberada como calor através de processos coletivamente conhecidos como decadência nãoraditória; e 4) o fóton é absorvido e a energia é rapidamente reemitida como um fóton secundário através do processo conhecido como fluorescência. Uma molécula cuja estrutura permite a emissão de fluorescência é chamada de fluorófora. A maioria das amostras biológicas contém fluoroforos endógenos insignificantes; portanto, fluoroforos exógenos devem ser usados para destacar características de interesse na amostra. Durante a microscopia de fluorescência, a amostra é iluminada com luz do comprimento de onda apropriado para absorção pelo fluoróforo. Ao absorver um fóton, diz-se que um fluoróforo está "animado" e o processo de absorção é chamado de "excitação". Quando um fluoróforo abre mão de energia na forma de um fóton, o processo é conhecido como "emissão", e o fóton emitido é chamado de fluorescência.
O feixe de luz usado para excitar um fluoróforo é focado pela lente objetiva de um microscópio e converge em um "ponto focal" onde é extremamente focado. Além do ponto focal, a luz novamente diverge. Os feixes de entrada e saída podem ser visualizados como um par de cones tocando no ponto focal (ver Figura 1, painel esquerdo). O fenômeno da difração impõe um limite de quão firmemente um feixe de luz pode ser focado - o feixe realmente se concentra em um ponto de tamanho finito. Dois fatores determinam o tamanho da mancha focal: 1) o comprimento de onda da luz, e 2) a capacidade de captação de luz da lente objetiva, que é caracterizada por sua abertura numérica (NA). O "ponto" focal se estende não só no plano x-y, mas também na direção z, e é na realidade um volume focal. As dimensões deste volume focal definem a resolução máxima alcançável por imagem óptica. Embora o número de fótons seja maior dentro do volume focal, os caminhos de luz cônica acima e abaixo do foco também contêm uma menor densidade de fótons. Qualquer fluoróforo no caminho da luz pode, assim, ser animado. Na microscopia convencional (campo largo), a emissão de fluoroforos acima e abaixo do plano focal contribuem com fluorescência fora de foco (um "fundo nebuloso"), o que reduz a resolução e o contraste da imagem, como demonstrado na Figura 1, com o cubo vermelho representando a emissão de fluorhoreo acima do plano focal (estrela vermelha) que resulta em fluorescência fora de foco (superior à direita). Este problema é amenizado em microscopia confocal, devido à utilização de um orifício. (Figura 2, inferior direito). Como descrito na Figura 3, o orifício permite que as emissões originárias do local focal cheguem ao detector (esquerda), enquanto bloqueiam a fluorescência fora de foco (direita) de atingir o detector, melhorando assim tanto a resolução quanto o contraste.

Figura 1. Resolução óptica de epifluorescência versus microscopia confocal. Clique aqui para ver uma versão maior desta figura.
O feixe de luz usado para excitar um fluoróforo é focado pela lente objetiva de um microscópio e converge em um volume focal e, em seguida, diverge (esquerda). A estrela vermelha representa o plano focal de uma amostra que está sendo imagen, enquanto o quadrado vermelho representa a emissão de fluoróforo acima do plano focal. Ao capturar uma imagem desta amostra usando um microscópio epifluorescente, a emissão do quadrado vermelho fora de foco será visível e contribuirá para um "fundo nebuloso" (canto superior direito). Os microscópios confocal têm um orifício que impede a detecção de luz emitida fora do plano focal, eliminando o "fundo nebuloso" (inferior direito).
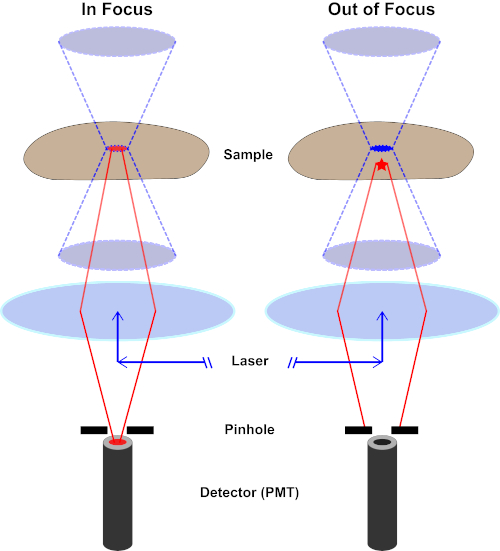
Figura 2. Efeito pinhole na microscopia confocal. Clique aqui para ver uma versão maior desta figura.
Embora a maior intensidade da luz de excitação esteja no ponto focal da lente (esquerda, oval vermelho), outras partes da amostra não no ponto focal (direita, estrela vermelha) receberão luz e fluoresce. Para evitar que a luz emitida dessas regiões fora de foco chegue ao detector, uma tela com um orifício está presente na frente do detector. Apenas a luz em foco (esquerda) emitindo do plano focal é capaz de viajar através do orifício e alcançar o detector. A luz fora de foco (à direita) é bloqueada com o orifício e não alcança o detector.

Figura 3. Principais componentes de um microscópio de varredura a laser confocal. Clique aqui para ver uma versão maior desta figura.
Por uma questão de simplicidade, a descrição mecanicista de um microscópio confocal será limitada à do Nikon Eclipse Ti A1R. Embora possa haver pequenas diferenças técnicas entre diferentes microscópios confocal, o A1R serve bem como um bom modelo para descrever a função de microscópio confocal. O feixe de luz de excitação, produzido por uma matriz de lasers de diodo, é refletido pelo espelho dicroico primário no objetivo, que foca a luz no espécime que está sendo imageado. O espelho dicroico primário reflete seletivamente a luz de excitação, permitindo que a luz em outros comprimentos de onda passe. A luz então encontra os espelhos de varredura que varrem o feixe de luz através do espécime de uma maneira x-y, iluminando um único (x,y) pixel de cada vez. A fluorescência emitida por fluoroforos no pixel iluminado é coletada pela lente objetiva e passa pelo espelhodicrómico primário para alcançar uma matriz de tubos fotomultiplier (PMTs). Espelhos dicroicos secundários direcionam a luz de emissão para o PMT apropriado. A luz de excitação espalhada pela amostra de volta ao objetivo é refletida pelo espelho dirítmico primário de volta para o espécime, e assim impedido de entrar no caminho da luz de detecção e chegar aos PMTs (ver Figura 3). Isso permite que a fluorescência relativamente fraca seja quantificada sem contaminação pela luz espalhada do feixe de luz de excitação, que é tipicamente ordens de magnitude mais intensas do que a fluorescência. Como o orifício bloqueia a luz de fora do volume focal, a luz que chega ao detector vem de um plano Zestreito e selecionado. Portanto, as imagens podem ser coletadas a partir de uma série de z-planesadjacentes; esta série de imagens é frequentemente referida como uma "pilha de z". Usando o software apropriado, uma pilha zpode ser processada para gerar uma imagem 3D do espécime. Uma vantagem particular da microscopia confocal é a capacidade de distinguir a localização subcelular da coloração. Por exemplo, a diferenciação entre a coloração da membrana a partir da coloração intracelular, que é muito desafiadora com a microscopia de epifluorescência convencional (1, 2, 3).
A preparação da amostra é uma faceta importante da imagem confocal. Uma força das técnicas ópticas de microscopia é a flexibilidade para imagem de células vivas ou fixas. Ao tentar produzir imagens 3D, devido ao número de imagens que devem ser adquiridas para uma pilha z, a dificuldade de manter a saúde celular, e o movimento de células vivas e suas organelas, o uso de células fixas é típico. O procedimento para fixação e coloração de células para fluorescência confocal é semelhante ao convencionalmente utilizado na imunofluorescência. Após a cultura em slides de câmara ou em deslizamentos de cobertura, as células são fixadas usando paraformaldeído para preservar a morfologia celular. A ligação de anticorpos não específicos é bloqueada usando albumina de soro bovino, leite ou soro normal. Para manter a especificidade dos anticorpos secundários, a solução utilizada não deve ser originária da mesma espécie em que os anticorpos primários foram gerados. As células são incubadas com anticorpos primários que ligam o antígeno de interesse. Ao rotular vários alvos celulares, os anticorpos primários devem ser derivados de uma espécie diferente. Anticorpos que marcam um antígeno são então ligados por anticorpos secundários conjugados por fluoróforo. Os anticorpos secundários conjugados por fluoróforo devem ser selecionados para que sejam compatíveis com os comprimentos de onda da excitação a laser disponíveis no microscópio confocal. Ao visualizar múltiplos antígenos, os espectros de excitação/emissão dos fluoroforos devem diferir o suficiente para que seus sinais possam ser discriminados pela análise microscópica. O espécime manchado é então montado em um slide para imagens. Um meio de montagem é usado para evitar fotobleaching e desidratação da amostra. Se desejar, um meio de montagem contendo uma contra-mancha nuclear (por exemplo, DAPI ou Hoechst) pode ser usado (4).
No protocolo a seguir, os fibroblastos de camundongos transfectados para expressar CD1d (LCD1) foram manchados com anticorpos que reconhecem CD1d e CD107a (LAMP-1). CD1d é um grande receptor do complexo de histocompatibilidade 1 (MHC 1) presente na superfície de células presentes de antígenos que apresentam antígenos lipídicos. LAMP-1 (proteína de membrana associada à linsósomal-1) é uma proteína transmembrana presente principalmente em membranas lysosômicas. Para apresentação adequada de antígeno, o CD1d é traficado através do compartimento lyososômico de pH baixo, por isso o LAMP-1 está sendo usado como um marcador do compartimento lysosomal para este protocolo. Ao sondar as células LCD1 com anti-CD1d e anti-LAMP-1 que foram produzidas em diferentes espécies, anticorpos secundários com fluoroforos únicos podem ser usados para determinar a localização de cada proteína na célula e se o CD1d está presente nos compartimentos lisesosmal positivos da LAMP-1.
Procedimento
1. Materiais
Buffers
- Tampão de lavagem: 1 X salina tamponada de fosfato estéril (PBS) sem cálcio ou magnésio
- Tampão de fixação: 1% paraformaldeído na PBS
- Tampão de permeabilização: 0,1% Triton X-100 na PBS
- Tampão de bloqueio: 1% de albumina de soro bovino na PBS
- Meio de crescimento celular: DMEM suplementado com 10% de soro bovino fetal (FBS), penicilina/estreptomicina e L-glutamina
Equipamento
- Capa de fluxo laminar
- Incubadora umidificada (37°C, 5% CO2)
- Microscópio de varredura a laser confocal; aqui, Nikon Eclipse Ti laser
Materiais e Reagentes
- Slides de cultura celular câmara
- Mídia de montagem anti-fade com DAPI (para núcleos de coloração)
- Vidro de tampa de microscópio
- Limpezas de tarefas delicadas
- Pipettors e dicas
Reagentes específicos de ensaio
- Células aderentes (células primárias ou linhas celulares); aqui, foram utilizados fibroblastos de camundongos transfectados com CD1d (LCD1).
- Anticorpos primários para detectar alvos celulares; aqui, foram utilizados CD107a anti-rato de rato (LAMP-1) e CD1d anti-rato do mouse.
- Anticorpos secundários conjugados por fluorofora específicos para isótipos de anticorpos primários; aqui igG anti-rato conjugado ao Alexafluor 488 e anti-mouse IgG conjugado ao Alexafluor 647 foram usados.
2. Protocolo
Preparando-se para a coloração de anticorpos
Células semeadas
- Resuspend as células de interesse em meios de crescimento.
- Em seguida, semente 500 μL da suspensão da célula /por poço nos poços de um slide de câmara de 4 poços. (Aqui, as células LCD1 foram semeadas em 2,5x105 células/câmara em 500 μL de mídia de crescimento. A densidade de semeadura pode variar entre as linhas celulares).
- Incubar o deslizamento da câmara durante a noite em uma incubadora de CO2 de 5%, a 37°C, para permitir que as células aderam ao vidro.
- No dia seguinte, aspire a mídia de cada poço e depois lave as células 1X com 500 μL PBS.
Fixação
- Para fixar as células, adicione 500 μL 1% de solução de paraformaldeído em cada poço e incubar por 15 minutos à temperatura ambiente.
- Após a incubação, colete o paraformaldeído em um recipiente de resíduos líquidos perigosos apropriado.
- Em seguida, lave as células 3 vezes com 500 μL PBS para remover quaisquer resquícios do fixador.
Permeabilização
- Permeabilize as células incubando com 500 μL de permeabilização tampão/poço por 15 minutos à temperatura ambiente.
- Em seguida, lave as células brevemente 3 vezes com 500 μL de PBS.
Bloqueio
- Incubar as células em cada poço com tampão de bloqueio de 500 μL por 1 hora a 4°C, para bloquear a ligação de anticorpos não específicos.
Incubação primária de anticorpos
- Aspire o buffer de bloqueio das câmaras de slides.
- Em seguida, adicione 500 μL de solução de anticorpos primários diluídos às células. (Aqui, o anti-CD107a (LAMP-1) foi diluído 1:500 e o anti-CD1d foi usado sem diluição (anticorpo monoclonal 1H6 foi gentilmente fornecido pelo Dr. Randy Brutkiewicz)).
- Incubar os slides durante a noite a 4°C.
Nota: Se sondar por mais de um alvo, certifique-se de que os anticorpos primários são isótipos diferentes. As concentrações recomendadas de anticorpos variam entre os fabricantes e devem ser tituladas antes do uso.
Incubação secundária de anticorpos
- Aspire a solução primária de anticorpos dos poços.
- Lave as câmaras de poço 4 vezes com 500 μL PBS.
- Em seguida, adicione 500 μL da solução de anticorpos secundários diluídos a cada poço. (Aqui, ambos os anticorpos secundários- anti-mouse IgG Alexafluor 647 e anti-rato IgG Alexafluor 488 foram diluídos 1:2000 no buffer de bloqueio).
- Incubar à temperatura ambiente por 1h no escuro.
- Após a incubação, aspire a solução de anticorpos secundários.
- Lave as câmaras 4 vezes com 500 μL PBS para remover qualquer anticorpo secundário não ligado.
Nota: As concentrações recomendadas de anticorpos variam entre os fabricantes e devem ser tituladas antes do uso. Se sondar por mais de um alvo, os anticorpos secundários devem ser conjugados a diferentes fluoroforos com espectros exclusivos de excitação/emissão. Tenha também em mente a excitação/emissão da contra-mancha nuclear (ou seja, DAPI) ao selecionar os fluoroforos. A seleção fluorófora pode ser impactada pela configuração a laser do microscópio confocal utilizado. A configuração a laser da máquina ditará quais fluoroforos são adequados para o experimento.
3. Deslizamentos de cobertura de montagem
- Primeiro, remova cuidadosamente as câmaras do slide.
- Em seguida, segure o slide no ângulo sobre uma delicada limpeza de tarefas e remova o fluido das bordas sem tocar nas células.
- Adicione 1 gota de meio de montagem antifato, contendo a mancha nuclear DAPI, em cada seção das células.
- Em seguida, coloque uma tampa de 20 mm x 60 mm sobre o slide, segurando-o sobre as bordas usando as pontas dos dedos. (Evite a formação de bolhas sobre as células, pois elas interferem com a imagem).
- Limpe qualquer meio de montagem extra nas laterais com uma delicada limpeza de tarefas e armazene os slides no escuro à temperatura ambiente até uma semana.
4. Imagem confocal
Amostras de imagem em um microscópio de varredura a laser confocal. Para dados mostrados na Figura 2, o Nikon Eclipse Ti A1R foi usado com o software NIS Elements Advanced Research. A seção a seguir detalha o procedimento para capturar imagens usando o software acima mencionado.
- Para começar a fotografar as células, abra o 'nis elements advanced research software' clicando no 'ícone de softwareNIS.'
- Em seguida, na janela de controle clique na guia'TiPad' e escolha o objetivo desejado para a imagem. (Aqui, foi utilizado o primeiro objetivo de 40x).
- Carregue o slide com células no palco e centralizar-o sob a lente.
- Agora, na aba 'A1plus Compact GUI', configure os lasers apropriados para os fluoroforos usados. Clique no símbolo de engrenagem para abrir o menu de configurações de corante e espectral e selecione os canais necessários e ajuste o laser para cada canal.
- Em seguida, selecione as emissões apropriadas no menu suspenso sob o primeiro espelhodicróico.
- Em seguida, sob a janela 'A1plus Compact GUI', clique em 'Ch. Series' para configurar a série de canais de linha, que configura se os lasers usados dispararão sobre a amostra simultaneamente ou sequencialmente. (Aqui, foram escolhidos passes sequenciais, começando pelo canal 1, seguido pelo canal 2, depois 4).
- Depois disso, comece a digitalizar clicando no ícone 'Ponta de seta' na parte superior. Neste ponto, enquanto a imagem estiver ao vivo, sob a janela' A1plus Compact GUI', clique na escala deslizante e modifique o tamanho do pinhole para garantir a limitação da luz de foco. (Aqui, foi utilizada a configuração mais baixa disponível (0,5).
- Em seguida, ajuste as configurações de 'alta tensão' e 'deslocamento' sob cada laser para níveis apropriados, usando as escalas deslizantes para permitir a detecção da coloração específica, limitando qualquer potencial coloração de fundo. Se uma amostra de coloração positiva estiver disponível, comece por imagem desta amostra para cada canal para garantir que as configurações de laser produzam as razões ideais de sinal para ruído.
Atenção: a alta intensidade do laser por longos períodos pode causar fotobleaching. - Depois de definir os valores ideais de HV e deslocamento para cada laser, clique na guia 'ND Acquisition' e, em seguida, selecione o ícone 'Z' para configurar os parâmetros para a série Z. Em seguida, ao adquirir uma imagem ao vivo da amostra, primeiro defina a parte inferior da imagem e clique no botão 'inferior' em seguida, encontrar a posição superior da amostra e clicar no botão 'superior'. Defina o tamanho da etapa digitando especificamente o tamanho da etapa preferencial em μm para cada etapa ou especificando quantas etapas totais são necessárias.
- Uma vez definidos os parâmetros da série Z, selecione a resolução de tamanho/pixel desejada da imagem. Para isso, clique na janela 'A1plus Compact GUI' e sob o ícone 'tamanho' selecione a resolução desejada. Para diminuir o ruído da imagem, pode-se selecionar o menu suspenso ao lado do símbolo 'ø' para obter uma média do número selecionado de imagens.
- Agora, clique na guia 'Executar agora' no menu 'ND Acquisition' para iniciar a imagem da amostra.
- Após a conclusão da imagem, salve a imagem clicando em 'Arquivo', então 'Salve Como', que exportará o arquivo de imagem com a extensão '.nd2'. Por fim, repita o processo para cada uma das outras amostras.
Resultados
Neste experimento, os fibroblastos de camundongos que expressavam o gene glicoproteína superficial CD1d foram corrigidos, imunossuídos e imagedos em um microscópio confocal. Uma imagem representativa obtida usando o protocolo acima é mostrada na Figura 4. No painel superior de A, imagens de um único canal mostrando o padrão de coloração de cada alvo individual são apresentadas. Essas imagens compreendem uma única seção (fatia) da pilha z capturada. O painel direito mostra a coloração da DAPI dos núcleos das células. Os painéis centrais mostram CD1d manchado em vermelho e LAMP-1, um marcador líssômico, manchado de verde. O painel esquerdo é uma imagem composta onde os três canais diferentes são mesclados. O aparecimento do amarelo resulta da sobreposição dos canais vermelho e verde, e indica uma área onde CD1d e LAMP-1 são co-localizados. Os resultados da coloração confirmam que o CD1d está localizado nos compartimentos endosomais LAMP-1+. Há também áreas onde apenas uma cor está presente, o que indica a presença de CD1d ou LAMP-1 sem co-localização. O painel inferior de A mostra uma renderização 3D das células construídas a partir de imagens capturadas na pilha z.
O painel B mostra uma fatia da pilha z na ampliação de 100x demonstrando os padrões de expressão dessas duas proteínas em maior detalhe. A caixa de delineado rosa no lado direito da imagem exibe a seção transversal da coordenada x designada pela linha rosa na imagem, que representa a vista lateral na linha rosa. Da mesma forma, a caixa de delineado azul na parte inferior da imagem mostra a seção transversal da coordenada y designada pela linha azul na imagem, que representa a vista frontal na linha azul. A renderização 3D da imagem z-stack permite que os usuários visualizem a imagem em 3D, visualizando todos os planos x, y e z.
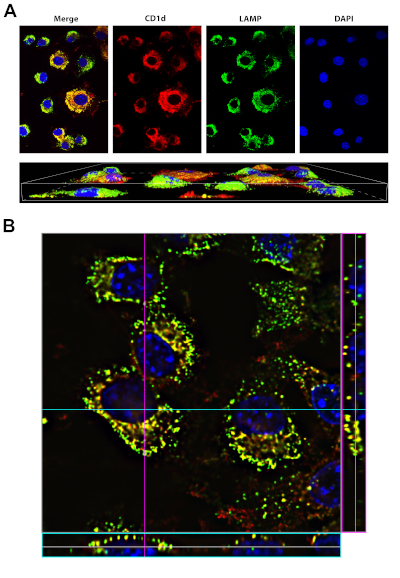
Figura 4: Mancha de CD1d e LAMP1. Clique aqui para ver uma versão maior desta figura.
A, painel superior: As células LCD1 foram fixadas, permeabilizadas e manchadas com anticorpos para CD1d (vermelho) e LAMP-1 (verde, um marcador do compartimento lysosomal). DAPI (azul, foi usado para visualizar o núcleo). A mesclagem (painel esquerdo) mostra que o CD1d está localizado no compartimento endosomal/lyossommal positivo LAMP-1 (amarelo).
A, painel inferior: renderização 3D das mesmas células no painel superior. As imagens foram adquiridas usando um objetivo de imersão em óleo de 40x no Nikon Eclipse Ti, usando o software NIS Elements Advanced Research.
B: imagem de 100x de células LCD1d manchadas como em A, com informações de pilha para uma determinada coordenada y (denotada pela linha azul) na parte inferior da imagem (caixa azul). As informações da pilha para uma determinada coordenada x (denotada pela linha rosa) são mostradas no lado direito da imagem (caixa rosa).
Aplicação e Resumo
A coloração fluorescente confocal é um procedimento relativamente simples que resulta em imagens de altíssima qualidade de espécimes que são preparados de forma semelhante à microscopia de fluorescência convencional. Resumindo, as amostras são fixas, permeabilizadas e bloqueadas. Anticorpos primários contra uma proteína ou proteínas de interesse são permitidos a ligar, então anticorpos secundários conjugados fluoróforos são usados para visualizar a coloração. A microscopia de fluorescência confocal tem aplicações em muitas áreas de pesquisa. Por exemplo, ao colorar marcadores de organelas subcelulares, juntamente com uma proteína de interesse, a microscopia confocal pode ser usada para determinar os locais subcelulares de diversas proteínas. Em comparação com a microscopia de fluorescência convencional, a imagem confocal pode distinguir mais efetivamente entre a superfície celular e a localização intracelular de uma proteína. Além disso, a imagem confocal também pode ser usada para determinar se duas proteínas co-localizam dentro da célula. Embora não esteja descrita neste protocolo, a microscopia de fluorescência confocal também pode ser realizada em células vivas para detectar alterações dinâmicas.

Vídeo 1: Vídeo criado no software NIS Elements Advanced Research, destacando a capacidade de mover-se através da renderização 3D das imagens. Clique aqui para ver este vídeo (Clique com o botão direito do mouse para baixar).
Referências
- Claxton, N. S., Fellers, T. J. and Davidson, M. W. Laser scanning confocal microscopy. Department of Optical Microscopy and Digital Imaging, National High Magnetic Field Laboratory, Florida State University, 37 p., Unpublished (2010). Available at- http://www.vertilon.com/pdf/PP6207.pdf.
- Ojcius, D. M., Niedergang, F., Subtil, A., Hellio, R. and Dautry-Varsat, A. Immunology and the confocal microscope. Research in Immunology, 147 (3),175-88 (1996).
- Paddock, S. W. and Eliceiri K. W. Laser scanning confocal microscopy: history, applications, and related optical sectioning techniques. Methods in Molecular Biology, 1075, 9-47 (2014).
- Hoff. F. How to prepare your specimen for immunofluorescence microscopy. Philipps University Marburg, Institute of Cytobiology and Cytopathology, Germany. (2015) Available at- http://www.leica-microsystems.com.
Pular para...
Vídeos desta coleção:

Now Playing
Microscopia confocal de fluorescência: uma técnica para determinar a localização de proteínas em fibroblastos de camundongos
Immunology
43.0K Visualizações

Citometria de Fluxo e Separação de Células Ativadas por Fluorescência (FACS): Isolamento de Linfócitos B Esplênicos
Immunology
92.5K Visualizações

Seleção de Células Ativadas por Magnetismo (MACS): Isolamento de Linfócitos T Tímicos
Immunology
22.7K Visualizações

Ensaios ELISA: Indireto, Sanduíche e Competitivo
Immunology
236.8K Visualizações

Ensaio ELISPOT: Detecção de Esplenócitos Secretores de IFN-γ
Immunology
28.3K Visualizações

Imunohistoquímica e imunocitoquímica: imageamento de tecidos via microscopia de luz
Immunology
78.5K Visualizações

Geração de anticorpos: produzindo anticorpos monoclonais usando hibridomas
Immunology
43.3K Visualizações

Microscopia de imunofluorescência: Coloração por imunofluorescência de secções de tecido embebidos em parafina
Immunology
53.6K Visualizações

Técnicas Baseadas em Imunoprecipitação: Purificação de Proteínas Endógenas Usando Esferas de Agarose
Immunology
87.4K Visualizações

Análise do Ciclo Celular: Avaliação da Proliferação de Células T CD4 e CD8 Após Estimulação Usando Coloração CFSE e Citometria de Fluxo
Immunology
24.1K Visualizações

Transferência de células adotivas: introduzindo esplenócitos de camundongos doadores para um camundongo hospedeiro e avaliando o sucesso via FACS
Immunology
22.1K Visualizações

Ensaio para Morte Celular: Ensaio de Liberação de Cromo para Avaliar a Capacidade Citotóxica
Immunology
151.4K Visualizações
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados