
Method Article
・ デ ・ ノボヤップ/タズによる体細胞の幹細胞の生成
要約
体 SCs の可用性は再生医療の重要な疾患モデル作製と SC プロパティへの洞察力を得るために。再プログラムする実験的戦略の紹介、体外が単一の転写コアクチベーター ヤップの一過性の発現によって彼らの対応する拡張幹/前駆細胞の細胞に成体細胞を区別します。
要約
ここでプライマリの分化した細胞とターン ヤップ転写因子の発現による同じ系統の幹細胞/前駆細胞 (SCs) にそれらを分離するためのプロトコルを提案する.この方法では、マウス乳腺の管腔の分化 (LD) 細胞はターン完全に差別化された膵外分泌細胞膵臓にダクトのようなまたヤップは、乳腺 SCs の分子・機能特性を表わす細胞に変換されます。前駆細胞。同様に、内因性、自然の scs ヤップ誘起幹様細胞 (「ySCs」) 最終的にとして展開できること organoid 文化長期in vitro における異所性ヤップ/タズ、のさらなる必要性なし ySCs、遺伝性自己更新 SC のような状態に恵まれています。
ここに示すプログラムし直す手順を生成し、分化細胞から始まって様々 な組織の源の前駆細胞の in vitroを展開する可能性を提供しています。Ex vivo体細胞の単純な拡張は、細胞・発生生物学研究一般、腫瘍発生のメカニズムの理解より多くの再生医療のための含意を持ってください。
概要
組織固有の体性幹細胞 (SCs) は、損傷後組織更新と修理のため重要です。簡単に分離し、限りなくの展開ex vivo体細胞 SCs 表す基礎研究と疾患モデルの SC 用だけでなく、潜在的な再生療法のための重要な問題する可能性。ただし、この方向の進歩は様々 な上皮臓器体外の SC 状態のキャプチャの難しさによって制限されており。確かに、いくつかの成体は、常駐 SCs が存在しない、または容易に利用できない可能性があります。 または、番号と再生の可能性は、老化や病気の条件によって侵食される可能性があります。単一の転写コアクチベーターの式を報告することによって、このギャップを埋める開始、2016 年にヤップ (はい関連タンパク質) やその密接に関連蛋白質 TAZ (PDZ モチーフと転写活性化)、終末分化した細胞に効率的な運用および分子に対応する組織特異 SCs1から見分けがつかない機能、拡張可能な非発癌性、自家細胞集団を作成します。パルス持続ヤップまたは数日間 TAZ 活動体 SCs の自己更新の外観を誘導するために十分です。これは、さらに異所性のヤップ/タズ1の表現なし携帯世代を通じて送信することができますもはや連続的な発現に依存する安定した状態です。プロトコルは乳腺とこれらの組織の分化した細胞から膵臓のde novo上皮幹細胞/前駆細胞を生成するために使用プロシージャの詳細をここで紹介します。この手順は、現在のリプログラミング/分化転換アリーナでブラック ボックスを塗りつぶします。これらの方向の主な取り組みは細胞へ誘導多能性幹細胞 (iPSC) 状態に、これらの胚の変換を中心としたところ確かとに多能性 SCs より分化した細胞。ただし、Ips、発癌性一度成体の完全かつ効率的な分化2プロトコルの開発の必要性を高める導入します。しかし、この分化ステップ可能な場合でもは長期的な拡張性、自己組織化と器官再作成電位の価格で来る。これらは、内因性の組織固有の SCs のみと現在記述されていたヤップ誘起 SCs (ySCs) の代表的な事実に、臓器再生のための必須の属性です。同様に、様々 な転写因子を生成もカクテルを使用して別に 1 つのセル型の直接分化転換は不可欠の潜在的な3増殖と幹細胞性を欠いているセルを区別されます。
ここで説明する手順はまた内因性 SCs が拡大することができ、分化前のヴィヴォ4最近導入された organoid 技術を活用します。ヤップ誘起 SCs は、内因性の SCs が存在しない実験、生物や病気の条件でも organoid 形成 SCs を生成するかもしれません。他の初期化の手順の違いでヤップによって与えられる細胞の可塑性の種類が生体組織に発生する SC のような状態に復帰の唯一の形式に対応していること、したいと思います。SC のような気質の買い戻しは組織の修復や発癌活性化5と関連付けられています。いくつかの成体組織の恒常性に不可欠なヤップや TAZ が再生、腫瘍の増殖培養1,6,7,8 体 SCs の拡大に必要不可欠、9,10、11,12
プロトコル
動物のすべてのプロシージャを行った私たちの機関のガイドラインを遵守することと OPBA と保健省によって承認
1. ヤップ誘発乳腺幹細胞 (yMaSCs) の生成
注: セクション 1 のすべてのメディア ・ ソリューションの組成は、表 1に指定されます。
- 主な乳腺細胞の分離
- 細胞文化のフードの下で準備: 使い捨てメス、ヒアルロニダーゼ ソリューション、解離中、溶血性ソリューション、並べ替えのソリューション、コラーゲン コート液は Ca2 +キレート ソリューション、洗浄媒体 #1 洗浄中 #2、当期ソリューション乳腺の 2次元培養培地、氷冷 HBSS/PS.
- 典型的な実験の犠牲にする 10 雌マウス (いずれかの CD-1 C57BL/6 ひずみ)、8-12 週齢頚部転位によって。解剖前に豊富な 70% エタノール溶液に腹部を消毒します。
- 腹部の皮膚に沿って Y 字切開と慎重にデュモン ピンセットで慎重に引き上げて腹膜から腺を分離する乳腺を解剖します。10 mL の氷で非細胞接着皿に切り裂かれた腺を置き冷たい HBSS/PS (各料理の 20 腺)、あらゆる皮膚の断片を引き継ぐことを確かめます。
- ティッシュ文化フードの下で新鮮な HBSS/PS の 10 mL で各腺を一度洗うし、空の非細胞接着皿 (各料理の 20 腺) に配置します。細胞がそれらに固執する傾向がある組織培養プレートを使用しない材料の重要な損失を引き起こしています。
- 細かく 1 mm3フラグメントの均質なミックスを取得するまでは、メスの乳腺をミンチします。目詰まりを避けるために、転送ピペッティング組織塊の分解および少なくとも 5 倍 50 mL の円錐管に懸濁液 25 mL の血清ピペットと解離培地 10 mL にそれぞれの料理からみじん切りにした組織を回復します。
注: 効率的な消化手順 1.1.5 で組織の適切なミンチは欠かせません。 - 連続的な活発な動揺と 37 ° C で 1 時間インキュベートします。1 時間後、ホモジネートをチェック、塊がまだ存在する場合 10 分でインキュベーションを延長します。室温で 5 分間 400 x g で消化組織スピンダウン、上澄みを廃棄します。溶血性溶液 3 mL で組織ペレットを再懸濁します、氷 3 分を孵化させなさい。
注: 厳密なタイミングはこの段階で非常に重要ですので、溶血はむしろ厳しい治療です。 - 10 ml の洗浄媒体 #1 のセルを洗って 5 分 400 x g で消化組織スピンダウン上澄みを廃棄します。10 mL の洗浄中 # 2 と 10 cm ティッシュの培養皿でプレートに組織ペレットを再懸濁します。細胞文化のインキュベーターで 37 ° C で 1 時間の料理を孵化させなさい。
注: この手順は線維芽細胞を培養皿に従う必要がありますの大部分の除去になります。 - 料理から細胞懸濁液を回復し、50 mL の円錐管に注ぐ。5 分間 400 x g でスピンし、上清を除去します。餌を Ca2 + 400 × g で 5 分でダウン回転するたびに、ソリューションをキレート 10 mL で 2 回洗浄します。0.25% の 5 mL にペレットを再懸濁しますトリプシン EDTA/37 ° C で 5 分間インキュベートし、
- 5 mL のトリプシン溶液の上に dispase 溶液を追加し、DNase 私 1μg/mL を補完します。DNA の塊の分解および、3 分ごとに揺れ、37 ° C で 10 分間インキュベートし 1 mL 先端から少なくとも 5 倍上下ピペットします。
- 40 μ m 携帯こし器を通って新しい 50 mL の円錐管に洗浄中 #2 の 10 mL とフィルターを追加します。スピン ・ ダウン 5 分 400 x g で細胞懸濁液とすべての液体を排除するようにして、上澄みを廃棄します。
- FACS による乳腺上皮細胞の浄化
- 10 μ L の追加によって抗体ミックスを準備林 (マウス系統抗体カクテル)、12 μ L 抗-CD326 (Ep CAM)、30 ng/mL、25 ng/mL, 10 ng/mL の最終濃度に 10 μ L 抗-CD61 の最終的な集中に 10 μ L 抗-CD49f の最終的な集中に、2.5 μ L 抗-(cd29)、2.5 ng/mL の最終的な集中に。
注:このステップを 1.3 から常に蛍光に分類された抗体の漂白を避けるために、暗闇の中で動作します。各造粒のマトリックス。 - (ペレットに 100 μ L の先端を浸すこと) によって少数の細胞を別々 のチューブにしてください。500 μ L の並べ替えの FACS 管内ソリューションで細胞を再懸濁します、FACS プロシージャのラベルのサンプルのまま氷の上。
- 洗浄媒体 #1 の 200 μ L でペレットを再懸濁します、抗体ミックスの 44.5 μ L を追加、ピペットの徹底的に、暗闇の中で氷の上で 30 分間インキュベートします。洗浄媒体 #1 の 10 mL の細胞懸濁液を希釈、スピン ・ ダウン 5 分 400 x g で、上澄みを廃棄します。
- 並べ替えのソリューションの 2 mL で細胞ペレットを再懸濁します、キャップ ストレーナー FACS tubeand を介してフィルターが (図 1 b) のように細胞の FACS 分離に進みます。多色の FACS プロトコルを実行する前に、それぞれの人にそれぞれの螢光色素の波及の可能性を修正することを確認します。細胞懸濁液を個別に各蛍光標識抗体をインキュベートし、補償行列を作成するためにすべてフルオロおよび単一カラー コントロールを介して、すべての検出器のスペクトル重複値を測定します。
注: この実験で 85 μ m ノズル搭載の並べ替えアイコンが採用されました。10 のメスのマウスからの一般的な準備を行えば、約 800,000 LD セル。FACS プロシージャ中に塊を形成する場合、二次ろ過に進みます。
- 10 μ L の追加によって抗体ミックスを準備林 (マウス系統抗体カクテル)、12 μ L 抗-CD326 (Ep CAM)、30 ng/mL、25 ng/mL, 10 ng/mL の最終濃度に 10 μ L 抗-CD61 の最終的な集中に 10 μ L 抗-CD49f の最終的な集中に、2.5 μ L 抗-(cd29)、2.5 ng/mL の最終的な集中に。
- 主な乳腺 LD 細胞の播種
- FACS プロシージャ中にコラーゲンとマルチも組織培養プレート「私コート液」をコート。37 ° C で 1 h、5% CO2細胞文化のインキュベーターで孵化させなさい。コーティング ソリューションを取り外して洗浄中 #1 めっき直前に洗います。
- 10 ml の洗浄ソリューション #1 の FACS プロシージャから回復した細胞を洗浄、スピン ・ ダウン 5 分 400 × g と上清を除去します。乳腺の 2次元培養培地 (500 μ L/ウェル) と扱われるコラーゲン プレート細胞ペレットを再懸濁します。48 時間を可能にする適切な細胞接着および広がり細胞文化のインキュベーターで座っている細胞ができます。
注: 10 雌マウスから典型的な並べ替え、マルチ 24 ウェルのウェル プレートの 6-8 の井戸を行えば最適な細胞密度 (100 000 細胞/ウェル)。
- YMaSCs (ヤップ誘発乳腺幹細胞) の誘導
注: このステップからすべてのプロシージャは BSL 2 条件下で実行する必要があります。- 乳腺コロニー メディアを準備します。新鮮な培地に細胞播種直前に基底膜マトリックスを追加します。
- ヤップ誘発乳腺幹細胞 (yMaSCs) の誘導、レンチ ウイルス感染による乳腺無血清の 2 つのボリュームと FUdeltaGW 情報を頼むことウイルス上清の 1 つのボリューム、FUW-重音テト-ヤップ (TAZ) の上澄みの 1 つのボリュームを混合してのプライマリ LD セルを変換する.2次元培養液 500 mL の容量で、サプリメントの 2 倍濃度。48 h のレンチ ウイルス上清を持つセルを孵化させなさい。一般的なレンチ ウイルスの準備オンライン プロトコル22を参照してください。
- 感染後付着性のセルを洗浄し、外因性のヤップ (TAZ) の遺伝子の発現を誘導するために 2 μ G/ml ドキシサイクリン添加乳腺 2次元培養液中で扱います。空、EGFP または YAPS94A の発現ベクトルまたは誘導のヤップ (TAZ) のベクトル, 感染細胞に感染細胞を使用せずネガティブ コントロールとしてドキシサイクリンが左。
注: 成功の感染は人間の YAP遺伝子の特定のプライマーで qRT PCR によって前述1として検証できます。 - ドキシサイクリンで誘導の 7 日後は、0.05% と孵化によって付着性のセルをデタッチ/トリプシン EDTA (150 μ L/ウェル) 37 ° C で 10 分間1:5 洗浄中 #2 (600 μ L/ウェル) を希釈して trypsinization を停止し、セルをカウントします。2 μ g/mL ドキシサイクリンと細胞/ウェル プレート 24 ウェル超添付ファイルで 1,000 のクローンの密度でシードを添加した乳腺コロニー中 (各ウェルの 1 mL)、細胞を再懸濁します。
注: 乳腺コロニー メディアが基底膜マトリックス添加時に冷たい氷であることを確認します。基底膜マトリックスの到着では、-20 ° C で保存する必要があります常にで、一晩ゆっくりと 4 ° C で融解解凍後、常に製造元のガイドラインによると、氷の上処理する必要があります。 - かつてヤップ-発現 LD 細胞増殖を開始 (播種後 14 日) 懸濁液 (yMaSC コロニー) の:masc のような植民地として成長、カウントおよび処理のさらなる分析 (図 1)。
注: (ポイント 1.4.3) のように陰性対照セルは、単一のセルとして保持されます。 - YMaSC コロニー成長; 14 日間新鮮な乳腺コロニー媒体ごとの 72 時間と文化を補充します。これを行うに 5% 基底膜マトリックス、サプリメントの 10 倍の濃度で添加することがなく乳腺のコロニー中の因数を準備し、1:10 を各ウェル (例えば100 μ L 総媒体の 1 mL の) の過度の希釈を避けるために容量の追加マトリックスのサスペンション。
- YMaSCs のサブ養殖
- 乳腺のコロニーの中から主な植民地を回復し、分離、シードします。
注: ヤップ再 LD 細胞由来 yMaSC 植民地自己再生能力を獲得、さらにドキシサイクリン投与 (すなわち、トランスジェニック ヤップ/タズの式とは関係なく) 正常にサブに培養することができます。 - 1.4.1 の手順と乳腺のコロニー中、乳腺における媒体を細胞培養のボンネットの下に準備します。
- 各サンプルを収集し、氷の余分なボリューム (10:1) で孵化させなさい冷たい HBSS 基底膜マトリックスを可溶化するために、氷の上に 1 時間。3 のコロニーを洗って 5 分と氷で再懸濁します 180 x g で遠心分離によって x 冷たい HBSS。植民地を 0.05% トリプシン EDTA/単一細胞懸濁液を取得する 37 ° C で 10 分間、インキュベートします。単一セルのレベルに完全な解離を確実に p1000 チップで 10 倍上下ピペット コロニー。
- ドキシサイクリン細胞/ウェル プレート 24 ウェル超添付ファイルで 1,000 のクローン密度でなく乳腺コロニー中 (各ウェルの 1 mL) の数とシードのセルします。自己更新を評価する手順をすべての 10-14 日継これを繰り返します。
- 3 つ目の通路の前に通路 yMaSC の植民地における培養条件、yMaSC の拡張性を高めるため、ミニ-腺、密接に生体内で乳腺を連想させる二層からなる上皮における自己組織化の形成を許可するには組織。
- 1.5.3 に 1.5.4 の手順と乳腺のコロニー中からの植民地を回復します。植民地を再懸濁します 100% 成長因子は、行列の 150 μ L で 24 ウェル超取り付けプレートの各ウェルに 20-25 コロニーの最大の replate を考慮した基底膜マトリックスを減少します。
- 37 ° C で 40 分間細胞文化のインキュベーターでプレートをインキュベートし、基底膜マトリックスを固めるし、乳腺における媒体の 500 μ L をゲルを重ねてください。
- 数日後、新進の organoids (図 1E) フォームにコロニーの形成を確認します。
- 後 10-14 日、道またはプロセスの詳細な分析 organoids。
- オルガノイド培養の通路、各試料を採取し、氷の過剰量 (10:1) に培養してオルガノイドを回復する冷たい HBSS 基底膜マトリックスを可溶化するために、氷の上に 1 時間。洗って organoids 3 スピン x 180 x g 5 分でダウンし、氷で再懸濁します冷たい HBSS。
- 0.05% トリプシン EDTA/単一細胞懸濁液を取得する 37 ° C で 10 分間でオルガノイドを孵化させなさい。単一セルのレベルに完全な解離を確実に p1000 チップで 10 倍上下オルガノイドをピペットします。
- 100% 成長因子減少の基底膜マトリックス (24 ウェル超取り付けプレートの各ウェルに 150 μ L) のドロップで単一細胞懸濁液として再シードします。基底膜マトリックス培養細胞文化のインキュベーターで 37 ° C で 40 分でゲルを形成し、乳腺における媒体の 500 μ L をゲルをオーバーレイをしましょう。
注: yMaSC organoids はステップ 1.5.13、trypsinization を避けることのように 100% 基底膜マトリックス文化からの回復によって凍結保存することができます。10 %dmso を添加した乳腺における記憶媒体に格納します。 - すぐに-80 ° c yMaSC オルガノイドを凍結し、液体窒素で保存します。
- 乳腺のコロニーの中から主な植民地を回復し、分離、シードします。
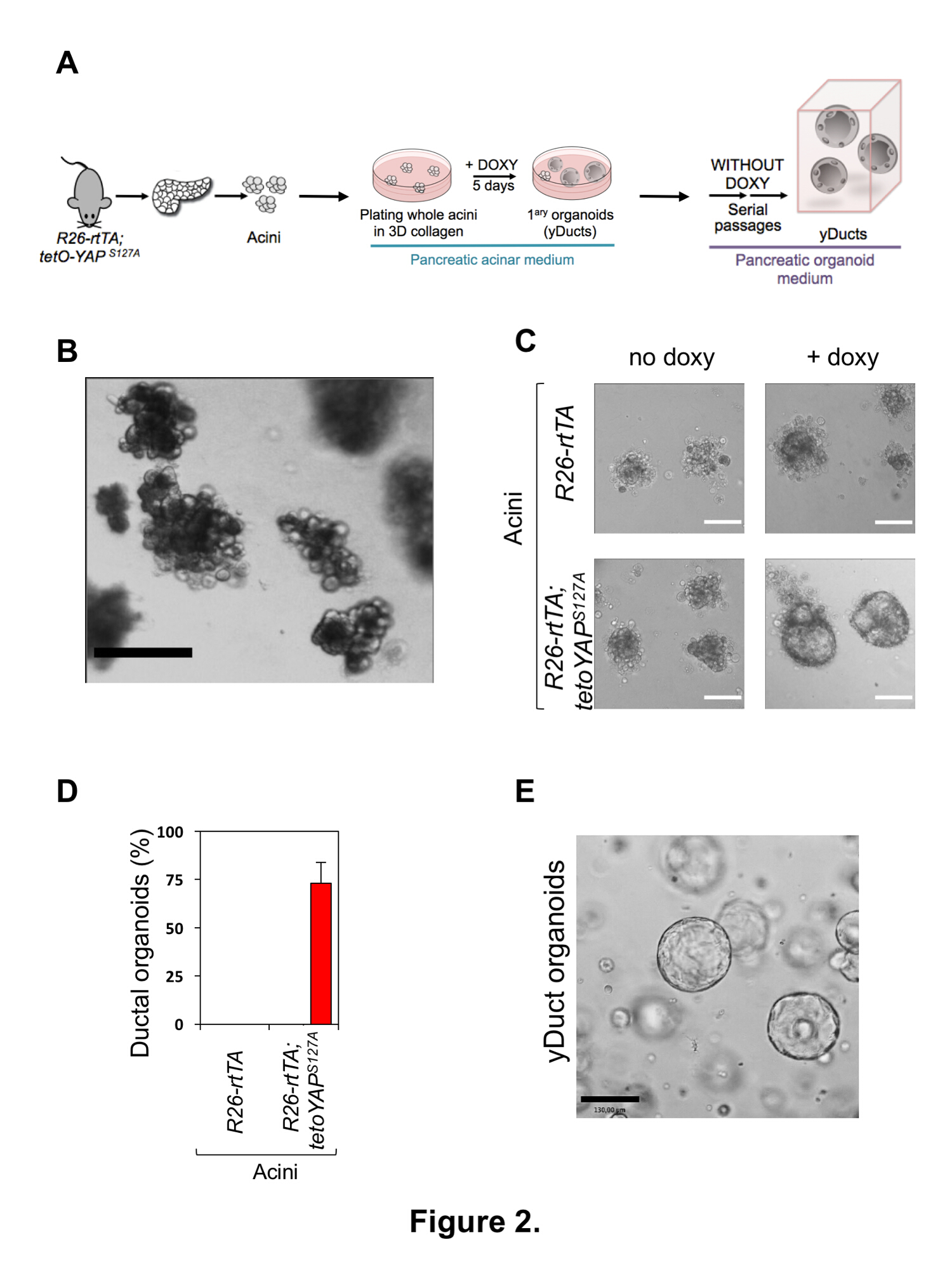
2. yDucts の生成
注: セクション 2 のメディア ・ ソリューションのすべての組成物は、表 2で指定されます。
- 主な膵腺管構造の分離
- 70% で解剖鉗子、はさみを置く EtOH セル フード腺房培養培地、各マウスの 15 mL の下の準備と腺房のリカバリ媒体、各マウスの 60 mLPBS/PS;原液コラゲナーゼ;コラゲナーゼ私溶液、各マウスの 15 mLラット尾コラーゲンを中和私ソリューション。ラット尾コラーゲンを中和する pH = 7、私とそれを中和するために氷の上のすべての試薬、ラット尾コラーゲン PBS/PS. 維持内コラーゲンが解散し、2.5 mg/mL に 10 n 塩酸 Dilute と酢酸のバッファーに 0.1 N NaOH で最初に調整することによって。適切な遺伝子型の 9 週齢のマウスに 6 を犠牲に。
- その裏に各マウスを置き、70% エタノール溶液による腹部を洗います。腹部の壁に沿って縦切開を確認します。(ガイドとしての脾臓を用いて) 膵臓を解剖し、10 mL 氷で 10 cm 非細胞接着お皿に冷たい PBS/PS. 転送細胞文化フードの下ですぐに料理。このステップからセル文化フードの下で常に動作します。
- 新しい非細胞各膵を転送接着以前がつまった一品コラゲナーゼの 7 mL 私ソリューション a.
- すぐに均質な組織懸濁液は約 1 mm3のフラグメントを取得する、使い捨てのメスのペアでそれぞれの膵臓をミンチします。
注: この手順が最適な細胞生存率の以上 2 分を取る必要があります注意してくださいすることが重要です。 - 37 ° C でコラゲナーゼの消化力の 5% CO2揺れ同種組織消化を確保するためにすべての 3 分, 10 分間細胞文化のインキュベーターで皿を孵化させなさい。
- 50 mL コニカル チューブ (各膵臓 1) 消化組織を回復、10 ml の培地洗浄腺房のお皿を洗って、同じ 50 mL の円錐管に配置、以上 3 倍上下組織を消化ピペッティングします。
- 18 ° c 100 × g で 5 分間消化組織スピンダウンし、上清を削除します。
注: この手順中にコラゲナーゼの活性を下げるための 18 ° C で細胞をスピンします。 - 組織を再懸濁しますコラゲナーゼの 7 mL にペレット私ソリューションは、このソリューションを新しいの 10 cm の非細胞接着を皿に入れて。
- ステップ 2.1.5、同種組織消化を確保するために 3 分ごとに揺れのように 10 分の 37 ° C でコラゲナーゼの消化力の第 2 ラウンドの皿を孵化させなさい。一方で、1 つのきれいな 50 mL の円錐管を 100 μ m の細胞のストレーナーをトッピング各膵臓に備えます。
- 消化組織を回復し、無菌の 10 mL シリンジのプランジャー (必ずストレーナー面に接線方向せん断力を回避、組織を慎重に押して) と組織の軟化で 100 μ m の細胞のストレーナー通過。腺房の洗浄中の 10 mL でお皿を洗って、同じ 100 μ m セル ストレーナーをこの 10 mL を通過します。
- 18 ° c 100 × g で 5 分間消化組織スピンダウンし、上清を削除します。
- 10 ml の腺房の洗浄中の組織ペレットを回復します。すでに追加の 10 mL の新鮮な腺房洗浄媒体を含む、ペレットの再懸濁のため過剰なピペッティングを回避 50 mL の円錐管に携帯ソリューションを転送します。
- 18 ° c 100 × g で 5 分間消化組織スピンダウンし、上清を削除します。
- 慎重に腺房の回復中期の 6 mL で消化組織を再懸濁します、6 もマルチも組織培養プレート、3 mL の 2 井戸でそれを配布します。顕微鏡下で慎重に評価する腺房の分離の質 (図 2B参照) 単一細胞のマイナーな割合と腺房のクラスターの均質な懸濁液として表示されます。任意の大きな組織を削除塊最終的に提示 (通常も肉眼に目に見える)、ソリューションからそれらをピペッティングします。
- 主な膵腺の播種
- 細胞の回復を許可する 2 h の携帯文化のインキュベーターで 37 ° C で消化腺房クラスターを孵化させなさい。
- 細胞回復コート中 48 ウェル マルチは私を中和ラット尾コラーゲン 100 μ l 井戸し、ハイドロゲル クッション フォームを許可する細胞文化のインキュベーターで 37 ° C で 1 時間インキュベートします。
- 細胞回復の 2 h は、円錐管の腺房細胞懸濁液を収集後、18 ° c 100 × g で 5 分間スピン ・ ダウンと上澄みを除去します。
- 腺房培 (48 よく組織培養プレートの各ウェルに 150 μ L) の適切なボリュームで、房を再懸濁します。各シードは、16 井戸最適密度 (100-120 腺房クラスター/も) を得るために膵臓を分離しました。
- 中和ラット尾コラーゲンの等量とこの腺房の懸濁液を希釈私ソリューションを氷の上の管を維持します。ミックス慎重かつ迅速に 2.2.2 (48 もマルチも組織培養プレートの各ウェルに 300 μ L) で説明されているコラーゲン クッションの上に細胞懸濁液をシードします。
- フォームにゲルを許可する細胞文化のインキュベーターで 37 ° C で 1 時間インキュベートします。
- 膵オルガノイドの誘導
- 500 μ L の腺房添加培養液中R26 rtTAM2; から膵オルガノイドのヤップ依存型誘導 2 μ g/ml ドキシサイクリンとコラーゲン ゲルをオーバーレイします。重音テト-ヤップS127Aマウス;マイナス コントロールが同じ条件やR26 rtTAM2; でwt細胞によって提供されます。重音テト-ヤップS127Aドキシサイクリンの不在で培養された細胞。
- 5 〜 7 日ごと 48 時間培養培地 (300 μ L/ウェル) を更新し、嚢胞形成性膵管のような構造 (への形態学的変化によって organoid 形成 2 μ g/mL ドキシサイクリンを添加した腺房細胞腺培養培地で図 2)。セルの膵臓における培養条件下で継代またはさらなる分析のために収穫できます organoids が形成される (例: RNA の抽出、蛍光抗体法)。
- サブの膵オルガノイド培養
- 自己複製能力を評価するためにクローン通路ヤップによる膵 organoids (yDucts) 三次元基底膜マトリックス ヒドロゲル (膵における培養条件) 外因性のヤップ/タズ供給 (すなわちとは無関係で。、ドキシサイクリン投与とは無関係)。
- トリプシンの準備 0,05%/EDTA。100% 成長因子減少の基底膜マトリックス;膵臓における媒体およびコラゲナーゼ私解決法 B
- 15 ml コニカル ソリューション B の継代するを各ウェルにコラゲナーゼの 4 mL と管します。
- 培を破棄、慎重に穏やかな吸引によって井戸からヒドロゲルを抽出し、円錐管にそれらを転送します。
- コラーゲン マトリックス (チェック、ハイドロゲルが完全に溶解するまで 10 分毎) の完全な消化力を許可する連続的な活発な動揺との 30 分のための 37 ° C でチューブを孵化させなさい。スピン ・ ダウン 2 分 750 x g で回復した細胞と上清を除去します。
- 孵化 1 ml のトリプシンの回復した細胞懸濁液を 1 つのセルを取得する 37 ° C で 10 分間 0,05%/EDTA。9 ml PBS 1 のトリプシンを希釈 x 750 x g 2 分でスピンダウンし、上清を除去。
- 超低接着プレート (通常 24 ウェル プレートのウェル 1 個で 150 μ L のドロップ) 種子および氷冷たい成長因子減少の基底膜マトリックス細胞ペレットを再懸濁します。
- 細胞文化のインキュベーター 37 ° C で 40 分でプレートの孵化によって固化し、膵臓における培地で重ねて基底膜マトリックス ハイドロゲルを聞かせて (各ウェルの 500 μ L)。yDucts は、7-10 日 (図 2 D) に嚢胞状 organoids として成長します。
- 氷冷 PBS 1 で基底膜マトリックスから削除されることができますさらに継オルガノイド x 30 分後に洗濯 3 回スピン ダウン 180 x g は 5 分、氷冷 PBS 1 濁で x 行列持ち越し効果を回避します。Organoids は、トリプシン 0.05% 単一細胞懸濁液を取得する 10 分間遠心分離し、新鮮な基底膜マトリックスの再シード、膵臓における媒体 (2.4.7-2.4.8) のようにかぶせています。
- yDuct オルガノイドをステップ 2.4.9、trypsinization を避けるように 100% 基底膜マトリックス文化からの回復によっての液体窒素中で凍結保存することができ、10 %dmso を添加した膵 Organoid 媒体に格納します。
- yDuct organoids は-80 ° C ですぐにフリーズ、その後、液体窒素で保存します。
結果
YMaSCs の生成
ヤップの発現による主な乳腺 LD 細胞を再プログラムする実験的戦略の概要については、図 1 aにされます。一次乳腺 LD の上皮細胞は13を並べ替え蛍光活性化細胞によって精製しました。図 1 bは、3 つの異なる集団を取得する典型的な並べ替えプロシージャを表します: 基底細胞 (EpCAM低CD49f高CD61-)、Luminal 前駆 (LP) のセル (EpCAM高CD49f低CD61+) と LD セル (EpCAM高CD49f低CD61-)。それが完全に区別される LD セルの純粋な調製を分離するために不可欠な 3 つの subpopulations のゲートは注意してくださいと成形条件 (参照図 1、左右パネル) 乳腺コロニーでシードするとき完全に成長が逮捕されました。逆に、外因性ヤップを表現する誘導、LD セルは 5% 基底膜マトリックス培養 (図 1) で簡単に認識できる密な上皮コロニーを形成する増殖を開始します。リプログラミング、効率は、約 3% の一般的な実験、単一細胞基底膜マトリックス培養 (図 1) でもともとシード数コロニー数をカウントすることによって獲得することができますを証明しました。プログラムし直された管腔細胞 (yMaSCs) 100% 基底膜マトリックスにおける培養条件 (図 1 aのスキームを参照してください)、複数ルーメン周辺開発複雑な器官毛細状構造に自己組織化への通路をすることができますし、ドキシサイクリン (すなわち遺伝子組換えヤップ発現の不在で) の不在でも驚くべき自己複製能力を表示 (図 1E)。病理 yMaSC 派生 organoids 表示基底層 (K14 正)、ECM と organoid (図 1 階) 内ルーメンのような空洞を直面している腔層 (K8 の肯定的な)、再構成した基底膜マトリックスに直面しています。このアーキテクチャは、ネイティブ MaSCs (図 1 f) によって形成されたオルガノイドの区別ではありません。
YDucts の生成
図 2 aで、ヤップの発現による主膵腺を再プログラムする実験的戦略を説明はします。全体の腺房のクラスターは、軽度の解離とろ過によりサイズ排除の組み合わせによる膵組織の大部分から分離されます。一般的な準備は、図 2 bに提示されます。分離、内分泌膵や膵管ツリーと単一のセルに最小限の解離フラグメントによって汚染なしとの均一なサイズの外分泌腺の腺房単位の懸濁液として腺房細胞のクラスターが表示されます。膵や膵管の破片による汚染は欠乏フィルタ (ステップ 2.1.10) 過酷な取り扱いのための徴候単一のセルに腺房のクラスターの不要な解離過度コラゲナーゼ処理またはバッファリングされていないアクティビティ追加 SBTI 治療によって抑制することができます組織が発表したタンパク質分解酵素の可能性があります。
図 2; の典型的な腺房リプログラミング実験を提示します。3 D コラーゲンの文化の 5-7 日以内-私はドキシサイクリンの存在下でゲルに基づく、容易にR26 rtTAM2/重音テト ヤップS127Aマウス由来膵腺管のようなクラスターに薄い撰 (我々 というyDucts)、拡大中央キャビティ中で増殖する上皮細胞の単層。典型的な実験のための 70% 程度である、リプログラミング効率は、シード房 (図 2 D) の合計数以上のダクトのようなクラスター数の得点を簡単に測定できます。陰性対照細胞、つまりR26 rtTAM2/+細胞またはR26 rtTAM2/重音テト ヤップS127Aセル左ドキシサイクリン、なし常として残っているされませんこれら文化の条件で以前に報告される1 ポスト mitotic の腺房クラスター ,14,15。プログラムし直された yDucts、マトリゲル ベース organoid 文化条件16に単一セルのレベルで継代するにすることができます (図 2 aのスキームを参照)、(すなわちドキシサイクリンの不在でも驚くべき自己複製能力を表示します。遺伝子組換えのヤップ発現の不在) で (図 2 e)。

図 1:主な乳腺 LD の単離細胞や乳腺の誘導は幹細胞です。(A)実験手順の概略を採用し、主な乳腺 LD セルをプログラムし直します。(B)代表 FACS プロット典型的な並べ替えプロシージャを示す LD セルを浄化します。i) 解離細胞は生きているセル (P1; 青) の前方および側面の散布によるとゲートします。ii) 人口 P1 は、さらにその林のプロフィールによるとゲート、: リネージュ陰性細胞 (P2; 灰色) の個体を選択すると、系列陽性造血細胞を除くiii) 人口 P2 と分かれています、EpCAM高(P3; 黄色 + 緑) EpCAM、低(P6; 赤) の集団。iv) P3 と P6 は、さらにゲート、CD61/CD49f のプロフィールによると 3 つの集団に: EpCAM低CD49f高CD61-基底細胞 (P7; 赤)、EpCAM高CD49f低CD61+ LP 細胞 (P8; 黄色)EpCAM高CD49f低CD61 と- LD セル (P9; 緑)。(C)画像が乳腺のコロニー中播種後 15 日乳腺のコロニーを形成する示された構造感染 LD 細胞の能力の説明されます。コロニー形成にヤップを表現する細胞ターン細胞、陰性対照細胞 (EGFP 感染) に対しのまま成長阻止された単一のセル。スケールバー = 50 μ m。(D) (C) のように、示された細胞の能力を形作っているコロニーの定量化。データ平均 + 標準偏差で、それぞれ六つの技術的な複製を持つ 5 つの独立した実験の代表であります。(E)ヤップ書き換え乳腺幹細胞様の細胞 (yMaSCs) ドキシサイクリンの不在での三次元 100% 基底膜マトリックス ハイドロゲルは新鮮な内における培養条件に 12 日後の代表的なイメージ。スケールバー = 100 μ m。(F)代表的な蛍光 K14 (緑) の基底のマーカーと K8 の内腔のマーカー (赤) オルガノイドから派生するイメージ示された細胞における培養条件に 12 日後。スケール バー = 10 μ m。この図は予想et al., 20161から再現します。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}

図 2: プライマリ膵腺房細胞, 膵前駆細胞の誘導の分離。(A)実験手順の概略を採用し、主な膵外分泌細胞をプログラムし直します。(B)分離手順 (ステップ 2.1.14) 直後後主膵腺の代表的なイメージ。腺房の準備は、単一のセルの最小の存在と腺房のクラスターの均一な懸濁液として表示されます。スケール バー = 400 μ m。(C)主膵腺管構造の代表的なイメージはR26 rtTAM2 (上部パネル) から派生したまたは R26 rtTAM2; 重音テト ヤップS127A (下方のパネル) マウス 3 D コラーゲン培養私と -ハイドロゲルを用いた 5 日間の有無ドキシサイクリン (愛人)、示される。のみプライマリ腺のヤップ表現はドキシサイクリン添加後嚢胞のような器官毛細として育つセルに変換します。スケール バー = 70 μ m。(D) (C) のように遺伝子組み換えのヤップ発現時に管オルガノイドを形成する膵臓腺の能力の定量化。データ平均 + 標準偏差で、4 つの技術的なレプリケートを実行、5 つの独立した実験の代表であります。(E)ドキシサイクリンの不在での三次元 100% 基底膜マトリックス ハイドロゲルは新鮮な 3 通路後ヤップ reprogramed 管のような細胞 (yDucts) の代表的なイメージ。スケール バー = 130 μ m。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}
| 主な乳腺細胞の分離 | |
| Ca2 +キレート ソリューション | 4 ° C でのストア |
| EDTA | 0.02 w/V |
| PBS | |
| コラーゲン コート液私 | |
| 酢酸 0.02N、pH 3,23 | |
| ラット尾コラーゲン (コーティング) | 1:50 |
| 当期のソリューション | 4 ° C でのストア |
| Dispase | 5 mg/ml |
| PBS | |
| 解離中 | |
| DMEM:F12 | |
| ヒアルロニダーゼ原液 | 400 U/mL |
| ペン/連鎖球菌 | 1 x |
| 原液コラゲナーゼ私 | 600 U/mL |
| 溶血性ソリューション | 4 ° C でのストア |
| NH4Cl ソリューション | 1 パーツ |
| TrisBase 20.6 グラム/L の | 9 パーツ |
| 7.2 pH を調整します。 | |
| HBSS/PS | 4 ° C でのストア |
| HBSS | |
| ペン/連鎖球菌 | 2 x |
| ヒアルロニダーゼ原液 | 0.2 μ m のフィルターは、4 ° C で保存 |
| ヒアルロニダーゼ ウシ精巣 (粉末) | 2,000 U/mL |
| ナトリウム リン酸バッファー 1 M pH7.3 | |
| NH4Cl ソリューション | T. amb を格納します。 |
| H2O | |
| NH4Cl | 7.1 グラム/L |
| 7.65 に pH を合わせなさい | |
| 並べ替えのソリューション | 0.2 μ m のフィルターは、4 ° C で保存 |
| BSA | 0.1% |
| EDTA | 1 mM |
| HEPES pH 7 | 25 mM |
| PBS | |
| 洗浄媒体 #1 | |
| DMEM/F12 | |
| ペン/連鎖球菌 | 1 x |
| 洗浄中 #2 | |
| DMEM/F12 | |
| 政府短期証券 | 5% |
| ペン/連鎖球菌 | 1 x |
| 乳腺の 2次元培養培地 | |
| DMEM/F12 | |
| 政府短期証券 | 2% |
| ヘパリン | 4 mg/mL |
| L-グルタミン | 1 x |
| マウス bFGF | 10 ng/mL |
| マウス EGF | 10 ng/mL |
| ペン/連鎖球菌 | 1 x |
| 誘導と yMaSCs の継 | |
| 乳腺のコロニー中 | |
| DMEM:F12 | |
| 政府短期証券 | 5% |
| ヘパリン | 4 μ G/ml |
| L-グルタミン | 1 x |
| マトリゲル (播種する前にすぐに追加) | 5% |
| マウス bFGF | 20 ng/mL |
| マウス EGF | 10 ng/mL |
| ペン/連鎖球菌 | 1 x |
| 乳腺における媒体 | |
| 高度な DMEM:F12 | |
| B27 | 1 x |
| GlutaMax | 1 x |
| ヘパリン | 4 μ G/ml |
| Hepes | 1 x |
| 人間の頭 | 100 ng/mL |
| マウス bEGF | 20 ng/mL |
| マウス EGF | 50 ng/mL |
| R Spondin 1 | 1 μ G/ml |
表 1: yMaSCs の生成します。すべての異なる文化メディアと一次乳腺 LD 細胞の分離と yMaSCs (セクション 1) の誘導に必要なソリューションの構成
| 主な膵腺管構造の分離 | |
| 腺房の培 | |
| BPE | 50 μ G/ml |
| BSA | 0.1% |
| デキサメタゾン | 1 μ G/ml |
| 政府短期証券 | 0.1% |
| その X | 1 x |
| ペン/連鎖球菌 | 1 x |
| SBTI | 0.2 mg/mL |
| Waymouth の媒体 | |
| 腺房洗浄中 | |
| BSA | 0.1% |
| ペン/連鎖球菌 | 1 x |
| RPMI 培地 | |
| SBTI | 0.2 mg/mL |
| 腺房のリカバリ媒体 | |
| 腺房の培 | |
| 政府短期証券 | 30% |
| コラゲナーゼ私溶液 | |
| 腺房洗浄中 | |
| 原液コラゲナーゼ私 | 360 U/mL |
| PBS/PS | 4 ° C でのストア |
| PBS (リン酸緩衝生理食塩水) | |
| ペン/連鎖球菌 | 1 x |
| 原液コラゲナーゼ私 | -20 ° C でストア |
| コラゲナーゼ、型私 (粉末) | 6,000 U/mL |
| PBS | |
| 膵オルガノイドの継 | |
| コラゲナーゼ私ソリューション B | |
| PBS 1 x | |
| 原液コラゲナーゼ私 | 240 U/mL |
| 膵臓における媒体 | |
| 高度な DMEM/F12 | |
| B27 | 1 x |
| ガストリン | 10 nM |
| 人間 FGF10 | 100 ng/mL |
| 人間の頭 | 100 ng/mL |
| マウス EGF | 50 ng/ml |
| N-アセチルシステイン | 1.25 mM |
| ニコチン酸アミド | 10 mM |
| ペン/連鎖球菌 | 1 x |
| R Spondin 1 | 1 mg/mL |
| SBTI | 0.2 mg/mL |
表 2: yDucts の生成します。すべての異なる文化メディアとプライマリ腺房細胞の誘導分離および yDucts (セクション 2) の通過に必要なソリューションの構成
ディスカッション
ここで1を既に報告されているヤップの一過性の発現によって対応する前駆細胞の細胞 (または ySCs) にさまざまな組織の前のヴィヴォ最終分化した上皮細胞をプログラムし直すためのプロトコルは提示します。我々 は 2 つの手順の詳細な: 1 つレンチウイルスベクターとウイルス感染を回避し、トランスジェニック ヤップ式の活用第 2 1 つを FACS 精製細胞のプログラムし直すことができます。各プロトコルを分離するための効率的な戦略を提示し、プライマリ分化細胞の培養と外因性を強制する戦略ヤップ・ デ ・ ノボ体性組織固有拡張可能な幹細胞 (参照を生成する分化した細胞の遺伝子発現図 1 aと2 aのスキーム)。
我々 は決して否定的なコントロールのサンプル (図 1および2 C) から任意の副産物を検出という事実によって示されるように分化した細胞の純粋な人口を効果的に紹介する分離戦略に分離を行った。
本研究で一次乳腺 LD 細胞のリプログラミング用レンチウイルスベクターはドキシサイクリン誘導、遺伝子発現のタイトなコントロールの可能性を提供します。これにより、オンにして外因性ヤップ オフで表現されます。特に注意は、リプログラミング効率の面で支障が出るこのすることができます過剰なウイルス力価の使用を避けることで配置必要があります。プライマリ腺房細胞の場合我々 は最小限の操作でヤップ依存型のリプログラミングを取得する完全に遺伝子組換え手法に切り替えました。この後者の戦略は孤立した腺房クラスター、やっとのことでレンチ ウイルスの感染を受けやすい、非常に壊れやすい主膵腺に特に適しています。形質転換戦略では、ドキシサイクリン依存性遺伝子発現のタイトなコントロールのレンチウイルスベクターの同じ利点を提供しています。また、主な膵腺と悪用される形質転換戦略は LD の乳腺細胞のウイルス誘導リプログラミングと比較してはるかに高いリプログラミング効率の付加的な利点を負いません。異なったティッシュから得られるセルに関連付けられている異なる組み込み可塑性、を超えて膵リプログラミングが多い可能性がありますに由来するすべての制服と自律のヤップ式に関連付けられた式の高効率explanted セルです。特に、外因性ヤップがもはや必要である ySCs (yMaSC のコロニーおよび yDucts) の生成後自己再生能力に影響を与えずに実証しました。YSCs 内因性ヤップ/タズを再アクティブ化して外因性ヤップ1オフになっているときの自己更新のためそれらを使用するためです。
YSCs は確かに分化した細胞から現れる遺伝的系統トレース検証1リプログラミング実験の起源の細胞を制御することによって概念を検証しました。
広範な評価ySCs のリプログラミング ヤップによる通常体 SCs1として私を生成することを示しています) トランスクリプトーム レベルで ySCs 表示ネイティブ SCs; 大規模な重複ii) ySCs の分化の能力を表示し、いつもの原点の自分の組織の id に制限長子孫を生成できます。iii) ySCs は非変形し、非発癌性と生体内移植します。
ここを維持し、文化を展開する手順を述べる yMaSCs と yDucts organoids として 100% 基底膜マトリックス ヒドロゲルに埋め込まれています。これらの条件は、自己組織化の未分化性文化の長期的なメンテナンスを確保する三次元 organoids に ySCs、下流解析のアプリケーションで集団を幹これらを展開するを有効にします。未知の理由のため、我々 は yMaSC を取得に失敗しましたを置くことによってオルガノイド感染における培養条件で直接 LD 細胞プラスチック培養プレートのドキシサイクリン治療後 7 日つまり、乳腺のコロニーの条件の中間の成長ステップが不可欠です。私たちの手でさらにネイティブ MaSCs 培養継前に乳腺のコロニーの条件が必要です。さらに、単一のセルにプライマリのコロニーを分離を避けるため organoid 培養条件にそのままコロニーを転送ではなく我々 が最も効率的な organoid 副産物が得られます。
における培養条件はまた、organoids 窒素風呂で凍結する前に細胞解離を避けること彼らの行列から回復される ySCs、凍結保存する可能性を与えることの利点を耐えます。
発表手順を再プログラミング ヤップの対応する組織幹細胞 (テストしましたが乳腺、膵、神経細胞を用いた) に異なる成体組織から派生した分化細胞型に変換できます1。Ips または他のリプログラミングの努力からの違いでヤップ/誘導 SCs に起源のティッシュの思い出を維持できます。注記のうち、体細胞の幹のような特性に恵まれている細胞分化解消、細胞運命の可塑性の唯一の形態、組織の損傷後など、創傷治癒の5,17をサポートする生体内で観察されたリプログラミング,18,19,20. ヤップと TAZ が主通常恒常性に不可欠であることは注目に値するが、複数組織11,21の組織の修復のために重要です。一貫してここでプログラムし直す手順の生理的な機能を持つヤップ/タズ示されている最近に成人の腸内細胞の転換を引き起こすことによって潰瘍性大腸炎患者のマウス ・ モデルにおける腸管再生に必要とされます。19胎児の腸の機能が表示されます修復上皮。ヤップ リプログラミングところ体外をキャプチャするために挑戦している状態の体性幹細胞を生成する手段を提供することによって現在の誘導細胞可塑性戦略が広がります。このアプローチでは、ひと由来細胞にも拡張が体細胞の未分化状態の研究への再生医療応用から広範な関連性があるし、体細胞の拡張のための in vitro細胞幹します。
開示事項
著者は競合する金銭的な利益を宣言しません。
謝辞
重音テト-ヤップS127Aマウス; の贈り物ありがとう f ・ カマルゴR26 rtTAM2マウス (株式 #006965)、ジャクソン研究所から購入しました。FACS 手順のヘルプについては、キアラ Frasson とジュゼッペ ・通奏低音をありがとうございます。この作品は AIRC 特別プログラム分子臨床腫瘍 ' 5 パーミル」によってサポートされ、AIRC PI-、S.P とエピジェネティクスの旗艦プロジェクト CNR Miur によって許諾する s. p.このプロジェクトは、欧州連合のホライゾン 2020年研究および革新プログラム (許諾契約 DENOVOSTEM 号 670126) の下で欧州研究会議 (ERC) からの資金を受けています。
資料
| Name | Company | Catalog Number | Comments |
| 10 mL sterile syringes | Rays | 10LC | |
| 100 mm cell strainer | Corning | 352360 | |
| 15 mL sterile conical tubes | Corning | 430052 | |
| 24-well ultra low attachment plates | Costar | 3473 | |
| 40 mm cell strainers | Corning | 352340 | |
| 48-well multiwell plates | Corning | 353078 | |
| 50 mL sterile conical tubes | Corning | 430290 | |
| 6-well multiwell plates | Corning | 353046 | |
| Advanced DMEM/F12 | Gibco | 12634028 | |
| B27 supplement (50x) | Gibco | 17504001 | |
| BPE | Gibco | 13028014 | |
| BSA | Sigma | A9418 | |
| Collagenase, type I | Sigma | 17018029 | |
| dexamethasone | Sigma | D4902 | |
| Dispase | Gibco | 1705-041 | |
| Disposable scalpels | Swann-Morton | 0503 | |
| DMEM/F12 | Gibco | 11320033 | |
| DMSO | Sigma | D2650 | |
| DnaseI | Roche | 11284932001 | |
| doxycycline hyclate | Sigma | D9891 | |
| EDTA | Sigma | E5134 | |
| Ethanol 100% | Sigma | 51976 | |
| FACS tubes (with strainer caps) | Falcon | 352235 | |
| FBS | Gibco | 10270106 | |
| FITC anti-mouse CD326 (Ep-CAM) | BioLegend | 118208 | |
| FUdeltaGW-rtTA | Addgene | #19780 | |
| FUW-tetO-EGFP | Addgene | #84041 | used as negative control |
| FUW-tetO-MCS | Addgene | #84008 | used as negative control |
| FUW-tetO-wtYAP | Addgene | #84009 | |
| FUW-tetO-YAPS94A | Addgene | #84010 | used as negative control (transcriptionally dead YAP mutant) |
| GlutaMax | Gibco | 35050061 | |
| HBSS | Gibco | 24020117 | |
| HCl | Sigma | 30721 | |
| heparin sodium salt | Sigma | H3149 | |
| HEPES buffer solution (1M) | Gibco | 15630-056 | |
| human R-Spondin1 (His Tag) | Sino Biological | 11083-H08H-5 | |
| Hyaluronidase from bovine testes | Sigma | H3506 | |
| ITS-X | Gibco | 51500056 | |
| K-14 antibody | Life Technologies | Ab7800 | |
| K-8 antibody | Life Technologies | Ab14053 | |
| L-Glutamine | Gibco | 25030081 | |
| Lin (allophycocyanin [APC] mouse lineage antibody cocktail) | BD Biosciences | 51-9003632 | |
| Matrigel® Growth Factor Reduced Basement Membrane Matrix, Phenol Red-Free | Corning | 356231 | |
| N-Acetylcysteine | Sigma | A9165 | |
| NaOH | J.T.Baker | 0402 | |
| NH4Cl | Sigma | A9434 | |
| Nicotinamide | Sigma | 72340 | |
| non-cell adhesive 10 cm dishes (sterile polystirol petri dish ø 94) | ROLL | 18248 | |
| PBS 10x | Euroclone | ECM4004XL | |
| PE Hamster Anti-Mouse CD61 | BD Biosciences | 553347 | |
| PE-Cy5 Rat Anti-Human CD49f | BD Biosciences | 551129 | |
| PE/Cy7 anti-mouse/rat CD29 Antibody | BioLegend | 102222 | |
| Pen/Strep (10,000 U/mL) | Gibco | 15140122 | |
| Rat Tail Collagen I (coating) | Sigma | 122-20 | |
| Rat Tail Collagen I for 3D culture | Cultrex | 3447-020-01 | |
| recombinant human FGF10 | Peprotech | 100-26 | |
| recombinant human Noggin | Peprotech | 120-10C | |
| recombinant murine EGF | Peprotech | 315-09 | |
| recombinant murine FGF basic (bFGF) | Peprotech | 450-33 | |
| RPMI 1640 medium | Gibco | 31870025 | |
| SBTI (Trypsin inhibitor from Glycine max) | Sigma | T6522 | |
| Tris BASE | Roche | 11814273001 | |
| Trypsin-EDTA 0,05% | Gibco | 25300054 | |
| Waymouth medium | Gibco | 31220023 |
参考文献
- Panciera, T. Induction of Expandable Tissue-Specific Stem/Progenitor Cells through Transient Expression of YAP/TAZ. Cell Stem Cell. 19 (6), 725-737 (2016).
- Bar-Nur, O. Lineage conversion induced by pluripotency factors involves transient passage through an iPSC stage. Nat Biotechnol. 33 (7), 761-768 (2015).
- Xu, J., Du, Y., Deng, H. Direct lineage reprogramming: strategies, mechanisms, and applications. Cell Stem Cell. 16 (2), 119-134 (2015).
- Clevers, H. Modeling Development and Disease with Organoids. Cell. 165 (7), 1586-1597 (2016).
- Blanpain, C., Fuchs, E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science. 344 (6189), 1242281 (2014).
- Bai, H. Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatology. 56 (3), 1097-1107 (2012).
- Cai, J. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 24 (21), 2383-2388 (2010).
- Elbediwy, A. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development. 143 (10), 1674-1687 (2016).
- Taniguchi, K. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature. 519 (7541), 57-62 (2015).
- Zhang, W. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal. 7 (324), ra42 (2014).
- Zanconato, F., Cordenonsi, M., Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 29 (6), 783-803 (2016).
- Azzolin, L. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell. 158 (1), 157-170 (2014).
- Stingl, J. Purification and unique properties of mammary epithelial stem cells. Nature. 439 (7079), 993-997 (2006).
- Means, A. L. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development. 132 (16), 3767-3776 (2005).
- Shi, G. Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene. 32 (15), 1950-1958 (2013).
- Huch, M. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 32, 2708-2721 (2013).
- Fernandez Vallone, V. Trop2 marks transient gastric fetal epithelium and adult regenerating cells after epithelial damage. Development. 143 (9), 1452-1463 (2016).
- Yanger, K. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 27 (7), 719-724 (2013).
- Yui, S. YAP/TAZ-Dependent Reprogramming of Colonic Epithelium Links ECM Remodeling to Tissue Regeneration. Cell Stem Cell. , (2017).
- Pan, F. C. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development. 140 (4), 751-764 (2013).
- Panciera, T., Azzolin, L., Cordenonsi, M., Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol. 18 (12), 758-770 (2017).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved