Séquençage de l'ARNr 16S : une technique basée sur la PCR pour l'identification d'espèces bactériennes
Vue d'ensemble
Source: Ewa Bukowska-Faniband1, Tilde Andersson1, Rolf Lood1
1 Département des sciences cliniques Lund, Division of Infection Medicine, Biomedical Center, Lund University, 221 00 Lund, Suède
La planète Terre est un habitat pour des millions d'espèces bactériennes, dont chacune a des caractéristiques spécifiques. L'identification des espèces bactériennes est largement utilisée dans l'écologie microbienne pour déterminer la biodiversité des échantillons environnementaux et la microbiologie médicale pour diagnostiquer les patients infectés. Les bactéries peuvent être classées à l'aide de méthodes de microbiologie conventionnelles, telles que la microscopie, la croissance sur des médias spécifiques, les tests biochimiques et sérologiques, et les tests de sensibilité aux antibiotiques. Au cours des dernières décennies, les méthodes de microbiologie moléculaire ont révolutionné l'identification bactérienne. Une méthode populaire est le séquençage du gène 16S ribosomal RNA (rRNA). Cette méthode est non seulement plus rapide et plus précise que les méthodes conventionnelles, mais permet également d'identifier les souches qui sont difficiles à cultiver en laboratoire. En outre, la différenciation des souches au niveau moléculaire permet la discrimination entre les bactéries phénotypiquement identiques (1-4).
16S rRNA se joint à un complexe de 19 protéines pour former une sous-unité 30S du ribosome bactérien (5). Il est encodé par le gène 16S rRNA, qui est présent et fortement conservé dans toutes les bactéries en raison de sa fonction essentielle dans l'assemblage de ribosome; cependant, il contient également des régions variables qui peuvent servir d'empreintes digitales pour des espèces particulières. Ces caractéristiques ont fait du gène 16S rRNA un fragment génétique idéal à utiliser dans l'identification, la comparaison et la classification phylogénétique des bactéries (6).
Le séquençage du gène rRNA 16S est basé sur la réaction en chaîne de polymérase (PCR) (7-8) suivie du séquençage de l'ADN (9). PCR est une méthode de biologie moléculaire utilisée pour amplifier des fragments spécifiques d'ADN à travers une série de cycles qui comprennent:
i) Dénaturation d'un modèle d'ADN à double brin
ii) Annealing des amorces (oligonucléotides courts) qui sont complémentaires au modèle
iii) Extension des amorces par l'enzyme de polymérase d'ADN, qui synthétise un nouveau brin d'ADN
Un aperçu schématique de la méthode est montré dans la figure 1.

Figure 1 : Aperçu schématique de la réaction de PCR. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Il y a plusieurs facteurs qui sont importants pour une réaction réussie de PCR, dont l'un est la qualité du modèle d'ADN. L'isolement de l'ADN chromosomique des bactéries peut être effectué à l'aide de protocoles standard ou de kits commerciaux. Une attention particulière doit être prise pour obtenir de l'ADN exempt de contaminants qui peuvent inhiber la réaction de PCR.
Les régions conservées du gène 16S rRNA permettent la conception de paires d'apprêt universels (un avant et un revers) qui peuvent se lier à la région cible et l'amplifier dans n'importe quelle espèce bactérienne. La région cible peut varier en taille. Alors que certaines paires d'amorces peuvent amplifier la plupart du gène 16S rRNA, d'autres n'amplifient que des parties de celui-ci. Des exemples d'amorces couramment utilisées sont présentés dans le tableau 1 et leurs sites de liaison sont représentés à la figure 2.
| Nom d'amorce | Séquence (5'-3') | Avant/revers | référence |
| 8F b) | AGAGTTTGATCCTGGCTCAG | en avant | -1 |
| 27F (en) | AGAGTTTGATCMTGGCTCAG | en avant | -10 |
| 515F (en) | GTGCCAGCMGCCGCGGTAA | en avant | -11 |
| 911R | GCCCCCGTCAATTCMTTTGA | revenir sur | -12 |
| en 1391R | GACGCGGTGTGTRCA | revenir sur | -11 |
| 1492R | GGTTACCTTGTTACGACTT GGTTACCTTGTTACGACTT GGTTACCTTGTTACGACTT GGTTA | revenir sur | -11 |
Tableau 1 : Exemples d'oligonucléotides standard utilisés dans l'amplification des gènes 16S rRNA a).
a) Les longueurs prévues du produit PCR générées à l'aide des différentes combinaisons d'amorces peuvent être estimées en calculant la distance entre les sites de liaison pour l'avant et l'amorce inverse (voir la figure 2), par exemple la taille du PCR produit utilisant la paire d'apprêt 8F-1492R est de 1500 bp, et pour la paire d'apprêt 27F-911R 900 bp.
b) également connu sous le nom de fD1

Figure 2 : Figure représentative de la séquence de rRNA 16S et des sites de liaison d'amorce. Les régions conservées sont colorées en gris et les régions variables sont remplies de lignes diagonales. Pour permettre la plus haute résolution, l'amorce 8F et 1492R (nom basé sur l'emplacement sur la séquence d'ARNr) sont utilisées pour amplifier la séquence entière, permettant le séquençage de plusieurs régions variables du gène. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Les conditions de cyclisme pour le PCR (c'est-à-dire la température et le temps requis pour que l'ADN soit dénaturé, annealed avec des amorces et synthétisé) dépendent du type de polymérase qui est utilisé et des propriétés des amorces. Il est recommandé de suivre les directives du fabricant pour une polymérase particulière.
Une fois le programme PCR terminé, les produits sont analysés par électrophoresis de gel d'agarose. Un PCR réussi donne une seule bande de taille prévue. Le produit doit être purifié avant le séquençage pour enlever les amorces résiduelles, les désoxyribonucleotides, la polymérase et le tampon qui étaient présents dans la réaction PCR. Les fragments d'ADN purifiés sont habituellement envoyés pour le séquençage aux services commerciaux de séquençage ; cependant, certaines institutions effectuent le séquençage de l'ADN dans leurs propres installations de base.
La séquence d'ADN est automatiquement générée à partir d'un chromatogramme d'ADN par un ordinateur et doit être soigneusement vérifiée pour la qualité, car l'édition manuelle est parfois nécessaire. Après cette étape, la séquence génétique est comparée aux séquences déposées dans la base de données rRNA 16S. Les régions de similitude sont identifiées, et les séquences les plus similaires sont livrées.
Procédure
1. Mise en place
- Tout en manipulant des micro-organismes, il est nécessaire de suivre de bonnes pratiques microbiologiques. Tous les micro-organismes, en particulier les échantillons inconnus, doivent être traités comme des agents pathogènes potentiels. Suivez la technique aseptique pour éviter de contaminer les échantillons, les chercheurs ou le laboratoire. Lavez-vous les mains avant et après la manipulation des bactéries, utilisez des gants et portez des vêtements de protection.
- Effectuer une évaluation des risques pour le protocole expérimental pour l'isolement de l'ADN génomique et la purification des produits PCR. Certains réactifs peuvent être nocifs!
- La culture pure est essentielle pour le séquençage de l'ARNr 16S. Avant de procéder à l'isolement de l'ADN génomique, assurez-vous que le matériau de départ est entièrement pur. Cela peut être fait par le placage de strie pour isoler les colonies individuelles. Ceux-ci peuvent être cultivés plus loin strié sur des assiettes individuellement, ou dans le bouillon, si nécessaire.
- Équipement de laboratoire requis :
- Cycler thermique pour PCR. La fonction du cycleur thermique est d'augmenter et de baisser la température selon un programme établi. Lors de la création du programme, il vous sera demandé d'entrer les valeurs de température et de temps pour chaque étape PCR ainsi que le nombre total de cycles.
- Système d'électrophoresis de gel d'Agarose. Il est utilisé pour séparer les fragments d'ADN en fonction de leur taille et de leur charge. Dans ce protocole, l'électrophorèse de gel d'agarose sera employée pour visualiser la qualité de l'ADN génomique et des produits de PCR isolés.
2. Protocole
Note: Le protocole démontré s'applique au séquençage du gène 16S rRNA à partir d'une culture pure de bactéries. Elle ne s'applique pas aux études métagénomiques.
-
Cultiver des bactéries pour l'isolement de l'ADN génomique (aDNc).
- Cultivez votre micro-organisme sur un support approprié. Les médias liquides et solides peuvent être utilisés dans cette étape. Choisissez les conditions qui donnent la meilleure croissance. Pendant la planification de l'expérience, gardez à l'esprit que les bactéries à croissance lente peuvent avoir besoin de plusieurs jours pour atteindre la phase de croissance tardive/ stationnaire. Dans ce protocole, Bacillus subtilis 168 a été cultivé dans le bouillon de lysogénie (LB) pendant la nuit dans un incubateur secouant réglé à 200 tr/min, 37 oC.
-
Isolement de l'ADN g.
- Si des bactéries étaient cultivées sur un milieu solide, gratter certaines cellules à l'aide d'une boucle stérile et les suspendre à nouveau dans 1 ml d'eau distillée.
- Si les bactéries étaient cultivées dans un milieu liquide, utilisez environ 1,5 ml d'une culture du jour au lendemain.
- Pelleter les cellules par centrifugation (1 minute, 12 000 - 16 000 g), enlever le supernatant, et utiliser les cellules pour l'isolement gDNA à l'aide d'un kit commercial ou de protocoles standard[p. ex. préparation totale de l'ADN du CTAB (13) ou extraction de phénol-chloroforme (14)]. Ici, un kit commercial a été utilisé pour isoler l'ADNc de 1,5 ml de B. subtilis 168 culture de nuit, OD600 - 1,5.
Note 1 : Pour certaines bactéries Gram-négatives cette étape peut être omise et remplacée par la libération simple de l'ADN des cellules par ébullition. Suspendre les granulés bactériens dans de l'eau distillée et incuber dans un bloc chauffant réglé à 100 oC pendant 10 minutes.
Note 2 : Les cellules bactériennes gram-positives sont difficiles à perturber. Il est donc recommandé de choisir une méthode d'isolement gDNA ou un kit qui est dédié à l'isolement de ce groupe de bactéries.
-
gDNA contrôle de la qualité.
- Vérifiez la qualité de l'ADN gdna isolé par électrophoresis de gel d'agarose. Tout d'abord, mélanger 5 lL de l'ADNg isolé avec 1 l de la teinture de chargement (6x), et charger l'échantillon sur un gel d'agarose de 0,8% qui contient un réactif de coloration de l'ADN.
- Chargez une norme de masse moléculaire et exécutez l'électrophorèse jusqu'à ce que le front de teinture atteigne le fond du gel.
- Une fois l'électrophoresis terminée, visualisez le gel sur un transilluminateur approprié (uv ou lumière bleue). gDNA apparaît comme une bande moléculaire élevée épaisse (au-dessus de 10 kb). Un exemple de la vérification de la qualité de l'ADNg est illustré dans la figure 3.
- Si l'ADNc passe le contrôle de la qualité(c'est-à-direla bande moléculaire élevée est présente et il y a peu ou pas de frottis de l'ADNc), diluez votre gDNA en série en étiquetant d'abord 3 tubes microcentrifugecommement comme suit : « 10x », « 100x » et « 1000x ».
- Pipette 90 l d'eau distillée stérile dans chacun des 3 tubes.
- Prenez 10 L de la solution gDNA et ajoutez-la au tube marqué "10x".
- Pipet l'ensemble du volume(c.-à-100 l) de haut en bas à fond pour s'assurer que la solution est mélangée uniformément. Ensuite, prenez 10 L de la solution de ce tube et transférez-le dans le tube marqué "100x".
- Mélanger comme décrit précédemment et répéter la même procédure en transférant 10 L de la solution du tube "100x" au tube "1000x". Ces dilutions seront utilisées comme modèle dans la réaction PCR.

Figure 3 : Agarose gel électrophoresis de gDNA isolé de Bacillus subtilis. Lane 1: M - marqueur de masse moléculaire (de haut en bas: 10000 bp, 8000 bp, 6000 bp, 5000 bp, 4000 bp, 3500 bp, 3000 bp, 2500 bp, 2000 bp, 1500 bp, 1000 bp, 1000 bp). Voie 2: gDNA - ADN génomique isolé de Bacillus subtilis. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
-
Amplification du gène 16S rRNA par PCR.
Note: Le protocole PCR ci-dessous est optimisé pour une polymérase d'ADN particulière et une paire d'apprêt 8F - 1492R (voir le tableau 1). L'optimisation du protocole est nécessaire pour chaque paire de polymérases et d'amorce.- Décongeler tous les réactifs sur la glace.
- Préparer le mix pcR master tel qu'indiqué dans le tableau 2. Étant donné que la polymérase d'ADN est active à température ambiante, la configuration de la réaction doit être effectuée sur la glace, c'est-à-dire que les tubes PCR et les composants de réaction doivent être maintenus sur la glace tout le temps. Préparer une réaction par échantillon d'ADNc get et une réaction pour un contrôle négatif. Le contrôle négatif est un mélange PCR sans le modèle gDNA et est utilisé pour s'assurer que les autres composants de la réaction ne sont pas contaminés.
Note: En cas d'échantillons multiples, un mélange maître est couramment préparé. Master mix est une solution contenant tous les composants de réaction à l'exception du modèle. Il permet d'omettre le tuyauterie répétitif, d'éviter les erreurs de tuyauterie et d'assurer une grande cohérence entre les échantillons. Pour préparer le mix principal, multipliez le volume de chaque composant (à l'exception du modèle d'ADN) par le nombre d'échantillons testés. Mélanger tous les composants dans le tube microcentrifuge et pipet tout le volume de haut en bas plusieurs fois. - Aliquot 49 l du mélange principal dans les différents tubes PCR.
- Ajouter un modèle de 1 L dans des tubes avec le mélange maître. Pour un contrôle négatif, ajoutez 1 l d'eau stérile. Pour s'assurer que les composants sont bien mélangés, épileflez doucement le mélange vers le haut et vers le bas 10 fois avec une pipette réglé à 30-50 'L.
- Définir la machine PCR avec le programme indiqué dans le tableau 3.
- Mettez les tubes dans la machine PCR et démarrez le programme.
- Une fois le programme terminé, examinez la qualité de votre produit PCR par électrophoresis de gel d'agarose.
- Une réaction PCR réussie à l'aide de la paire d'amorces 8F-1492R donne une seule bande d'environ 1,5 kb (Figure 4). Si d'autres bandes (c.-à-d. des produits non spécifiques) sont présentes, optimisez le programme PCR en ajustant la température d'annealing. Si une seule bande de taille prévue est présente, passez à l'étape suivante. Ici, la réaction pcR avec 100x modèle dilué gDNA a donné le meilleur produit car il avait une bande forte de taille prévue et manquait de produits non spécifiques. C'est pourquoi il a été choisi pour être purifié et envoyé pour le séquençage.
- Avant le séquençage, le produit doit être nettoyé à partir d'amorces résiduelles, de désoxyribonucleotides, de polymérase et de tampon qui étaient présents dans la réaction PCR. Les produits PCR peuvent être isolés à l'aide d'un kit commercial de purification PCR. La réaction PCR est chargée sur une colonne qui contient une matrice de liaison de l'ADN. Le produit PCR se lie à la colonne, tandis que d'autres composants circulent dans la colonne. La colonne est ensuite lavée à l'aide d'un tampon de lavage, et enfin, l'ADN est élucidé dans le tampon de choix. Confirmez que le tampon d'élution qui est complété par le kit est compatible avec le séquençage.
- Envoyer le produit PCR purifié pour le séquençage de l'ADN. Suivez les lignes directrices pour la soumission d'échantillons de séquençage à l'installation de séquençage choisie. Pour la meilleure couverture de séquence, utilisez les amorces d'amplification PCR (les mêmes que dans la section 2.4.1) comme amorces de séquençage. Ici, les amorces 8F et 1492R ont été utilisées pour le séquençage du produit PCR.
| composant | Concentration finale | Volume par réaction | Volume par x réactions (mix master) |
| tampon de réaction 5x | 1x 1x | 10 l | 10 l x |
| 10 mM dNTP | 200 m | 1 l | 1 l x |
| 10 M Amorce 8F | 0,5 M | 2,5 l | 2,5 l x |
| 10 M Amorce 1492R | 0,5 M | 2,5 l | 2,5 l x |
| Polymérase de phusion | 1 unité | 0,5 l | 0,5 l x |
| Template ADN | - | 1 l | - |
| ddH2O | - | 32,5 l | 32,5 l x |
| Volume total | 50 l | 49 l x |
Tableau 2 : composants de réaction PCR. - utiliser l'ADNc dilué 10x, 100x ou 1000x à partir de l'étape 2.3.
| pas | température | temps | Cycles |
| Dénaturation initiale | 98oC | 30 sec | |
| Dénaturation | 98oC | 10 sec | 25-30 |
| Recuit | 60oC | 30 sec | |
| prolongation | 72oC | 45 sec | |
| Prolongation finale | 72oC | 7 min | |
| tenir | 4oC | ∞ |
Tableau 3 : Programme PCR pour l'amplification du gène 16S rRNA.

Figure 4 : Électrophoresis de gel d'Agarose des produits de PCR amplifiés utilisant des amorces 8F et 1492R et gDNA comme modèle. L'échantillon d'ADNc de B. subtilis (voir la figure 3) a été dilué 10, 100 et 1000 fois afin de tester les meilleurs résultats. Voie 1: M - marqueur de masse moléculaire (de haut en bas: 10000 bp, 8000 bp, 6000 bp, 5000 bp, 4000 bp, 3500 bp, 3000 bp, 2500 bp, 2000 bp, 1500 bp, 1000 bp, 750 bp, 500 bp, 250 bp). Voie 2: Réaction PCR avec le modèle dilué 10x. Voie 3: Réaction PCR avec le modèle dilué 100x. Voie 4: Réaction PCR avec le modèle dilué 1000x. Voie 5: (C-) - contrôle négatif (réaction sans modèle d'ADN). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
3. Analyse des données et résultats
Note: Le produit PCR est séquencé à l'aide des amorces avant (ici 8F) et inverses (ici 1492R). Par conséquent, deux ensembles de séquences de données sont générés, l'un pour l'avant et l'autre pour l'amorce inversée. Pour chaque séquence, au moins deux types de fichiers sont générés : i) un fichier texte contenant la séquence d'ADN et ii) un chromatogramme d'ADN, qui montre la qualité de la séquence.
- Pour l'apprêt avant, ouvrez le chromatogramme et examinez attentivement la séquence. Un chromatogramme idéal pour une séquence de qualité devrait avoir des pics espacés uniformément et peu ou pas de signaux de fond (Figure 5A).
- Si le chromatogramme n'est pas de haute qualité, la séquence doit être jetée, ou le fichier texte de séquence doit être révisé en fonction des éléments suivants :
- La présence de doubles pics dans tout le chromatogramme indique la présence de plusieurs modèles d'ADN. Cela peut être le cas si la culture bactérienne n'était pas pure. Une telle séquence doit être écartée (Figure 5B).
- Un chromatogramme ambigu pourrait résulter de la présence de différents pics colorés au même endroit. L'une des erreurs les plus courantes est la présence de deux pics de couleur différents dans la même position et l'attribution inappropriée des bases par le logiciel de séquençage (Figure 5C). Corrigez manuellement les nucléotides mal assignés et modifiez-les dans le fichier texte.
- Les chromatogrammes à basse résolution peuvent entraîner des « pics larges » qui causent souvent un mauvais comptage des nucléotides dans ces régions (figure 5D). Cette erreur est difficile à corriger, et donc les inadéquations possibles dans l'étape d'alignement supplémentaire ne doivent pas être traitées comme fiables.
- Une mauvaise qualité de lecture du chromatogramme et la présence de pics multiples sont généralement observées aux extrémités de 5' et 3' de la séquence. Certains logiciels de séquenceur suppriment automatiquement ces fragments de mauvaise qualité (Figure 5E), et les nucléotides ne sont pas inclus dans le fichier texte. Si votre séquence n'a pas été tronquée automatiquement, déterminez les fragments de mauvaise qualité(par exemple signal faible, pics qui se chevauchent, perte de résolution) aux extrémités et supprimez les bases respectives du fichier texte.
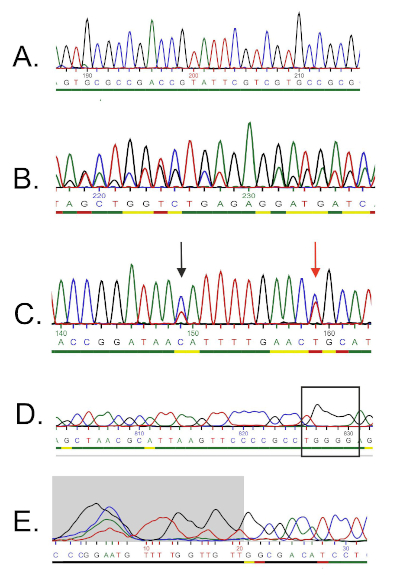
Figure 5 : Exemples de dépannage du séquençage de l'ADN. A) Exemple d'une séquence de chromatogrammes de qualité (des pics uniformément espacés et sans ambiguïté). B) Séquence de mauvaise qualité qui se produit habituellement au début du chromatogramme. La zone grise est considérée comme de mauvaise qualité et automatiquement supprimée par le logiciel de séquençage. Plus de bases peuvent être coupées manuellement. C) Présence de double pics (indiqués par des flèches). Un nucléotide indiqué par la flèche rouge a été lu par le séquenceur comme "T" (pic rouge), mais le pic bleu est plus fort, et il peut également être interprété comme "C". D) Les pics de chevauchement indiquent une contamination par l'ADN(c.-à-d. plus d'un modèle). E) Perte de résolution et soi-disant « pics larges » (marqués par rectangle) qui empêchent l'appel de base fiable. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
- Répétez 3.1 et 3.2 pour l'amorce inversée.
- Enfin, assemblez les séquences avant et inverses en une séquence contigu. Une bonne exécution de séquençage donne une séquence de jusqu'à 1100 bp. Considérant que le produit PCR est de 1500 bp de long, les séquences obtenues à l'aide d'amorces avant et inverses devraient se chevaucher partiellement.
- Fusionner les deux séquences à l'aide du programme d'assemblage de séquences d'ADN, par exemple un outil gratuit comme CAP3 (http://doua.prabi.fr/software/cap3) (15).
- Insérez les deux séquences en format EXPRES dans la case indiquée. Cliquez sur le bouton "Soumettre" et attendez que les résultats reviennent.
- Pour afficher la séquence assemblée appuyez sur "Contigs" dans l'onglet résultat. Pour voir les détails de l'alignement appuyez sur "Détails d'assemblage".
Note 1 : Si le logiciel CAP3 est utilisé pour l'assemblage contig, il n'est pas nécessaire de convertir la séquence d'apprêt inverse en inverse-complémentaire; toutefois, cette étape peut être nécessaire si un autre programme est utilisé.
Note 2 : Le format FASTA est un format textuel pour représenter la séquence de nucléotide. La première ligne (la ligne de description) dans un fichier FASTA commence par un symbole « 'gt;» suivi du nom ou d'un identifiant unique de la séquence. Suivant la ligne de description est la séquence de nucléotide. Collez vos séquences dans le format suivant :
'gt;séquence'frw'primer
coller votre séquence à partir du fichier texte ici
'gt;séquence'rvs'primer
coller votre séquence à partir du fichier texte ici
- Effectuez une recherche dans une base de données en visitant le site Web pour l'outil de recherche local de base (BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi).
- Sélectionnez l'outil "Nucléotide BLAST" pour comparer votre séquence à la base de données.
- Entrez votre séquence (le contig assemblé en 3.5) dans la boîte de texte "Séquence requête", puis sélectionnez la base de données "16S rRNA sequences (Bacteria et Archea)" dans le menu défilement vers le bas.
- Appuyez sur le bouton "BLAST" au bas de la page. Les séquences les plus similaires seront retournées. Un exemple de résultat BLAST est affiché dans la figure 6. Dans l'expérience présentée, le coup principal est la souche 168 de B. subtilis, montrant l'identité à 100 % avec la séquence disponible dans la base de données BLAST.
- Si le coup supérieur ne montre pas l'identité à 100%, allez à l'alignement et vérifiez les décalages. En cliquant sur le coup supérieur, vous serez dirigé vers les détails de l'alignement. Les nucléotides alignés seront rejoints par de courtes lignes verticales tandis que les nucléotides dépareillés ont un espace entre eux. Retournez au chromatogramme que vous avez reçu de la société de séquençage et révisez la séquence une fois de plus en mettant l'accent sur la région dépareillée. Corriger la séquence si des erreurs supplémentaires sont trouvées. Exécutez BLAST à nouveau en utilisant la séquence corrigée.
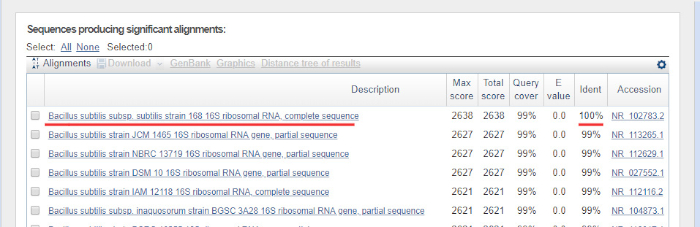
Figure 6 : Exemple du résultat de blast nucléotide. La séquence de gène de rRNA 16S de la culture pure de B. subtilis 168 a été employée comme séquence de requête. Le top hit montre 100% d'identité (souligné) à la souche B. subtilis 168, comme prévu. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Applications et Résumé
L'identification des espèces bactériennes est importante pour les différents chercheurs, ainsi que pour ceux dans les soins de santé. Le séquençage 16S de rRNA a été au commencement employé par des chercheurs pour déterminer des relations phylogénétiques entre les bactéries. Avec le temps, il a été mis en œuvre dans le cadre d'études métagénomiques visant à déterminer la biodiversité des échantillons environnementaux et dans les laboratoires cliniques comme méthode permettant d'identifier les agents pathogènes potentiels. Il permet une identification rapide et précise des bactéries présentes dans les échantillons cliniques, facilitant un diagnostic plus précoce et un traitement plus rapide des patients.
References
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.
Tags
Passer à...
Vidéos de cette collection:

Now Playing
Séquençage de l'ARNr 16S : une technique basée sur la PCR pour l'identification d'espèces bactériennes
Microbiology
188.0K Vues

Création d'une colonne de Winogradsky : une méthode pour enrichir les espèces microbiennes dans un échantillon de sédiments.
Microbiology
128.4K Vues

Dilutions en série et ensemencement des géloses : numération des micro-organismes
Microbiology
314.2K Vues

Cultures enrichies : Cultiver des micro-organismes aérobies et anaérobies dans des milieux sélectifs et différentiels
Microbiology
131.8K Vues

Cultures pures et ensemencement des géloses : isolement des colonies bactériennes pures à partir d'un échantillon mixte
Microbiology
165.7K Vues

Courbes de croissance : Générer des courbes de croissance en comptant les unités formant colonies (UFC) et en mesurant l'absorbance
Microbiology
293.4K Vues

Tests de sensibilité aux antibiotiques : Utilisation du ETEST pour déterminer la CMI de deux antibiotiques et évaluer la synergie des antibiotiques
Microbiology
93.5K Vues

Microscopie et coloration : Gram, Capsule et endospores.
Microbiology
362.5K Vues

Test de la plaque : méthode de détermination de la charge virale exprimée en unités formant des plaques
Microbiology
185.7K Vues

Transformation des cellules E. coli en utilisant le chlorure de calcium
Microbiology
86.3K Vues

Conjugaison : méthode de transfert de la résistance à l'ampicilline du donneur à l'hôte E. coli
Microbiology
38.1K Vues

Transduction via bactériophage : méthode de transfert de la résistance à l'ampicilline du donneur au receveur E. coli
Microbiology
28.9K Vues
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.