
Microscopie confocale à fluorescence : une technique pour localiser les protéines dans les fibroblastes de souris
Vue d'ensemble
Source: Dominique R. Bollino1, Eric A. Legenzov2, Tonya J. Webb1
1 Département de microbiologie et d'immunologie, University of Maryland School of Medicine et Marlene and Stewart Greenebaum Comprehensive Cancer Center, Baltimore, Maryland 21201
2 Center for Biomedical Engineering and Technology, University of Maryland School of Medicine, Baltimore, Maryland 21201
La microscopie par fluorescence confocale est une technique d'imagerie qui permet une résolution optique accrue par rapport à la microscopie d'épifluorescence « à champ large » classique. Les microscopes confocals sont capables d'obtenir une meilleure résolution optique x-y grâce à la « numérisation laser » - généralement un ensemble de miroirs à tension contrôlée (miroirs de galvanomètre ou de « galvo ») qui dirigent l'éclairage laser à chaque pixel du spécimen à la fois. Plus important encore, les microscopes confocals atteignent une résolution z-axiale supérieure en utilisant un sténopé pour enlever la lumière de mise au point provenant d'endroits qui ne sont pas dans le z-plan étant numérisé, permettant ainsi au détecteur de recueillir des données à partir d'un z-plan spécifié. En raison de la haute résolution Z réalisable dans la microscopie confocale, il est possible de recueillir des images à partir d'une série de z-planes (également appelé z-stack) et de construire une image 3D à travers un logiciel.
Avant de discuter du mécanisme d'un microscope confocal, il est important de considérer comment un échantillon interagit avec la lumière. La lumière est composée de photons, des paquets d'énergie électromagnétique. Un photon empiéchant sur un échantillon biologique peut interagir avec les molécules qui composent l'échantillon de l'une des quatre façons : 1) le photon n'interagit pas et passe à travers l'échantillon; 2) le photon est réfléchi/dispersé; 3) le photon est absorbé par une molécule et l'énergie absorbée est libérée sous forme de chaleur par des processus collectivement connus sous le nom de carie non radiative; et 4) le photon est absorbé et l'énergie est alors rapidement réémise en tant que photon secondaire par le processus connu sous le nom de fluorescence. Une molécule dont la structure permet l'émission de fluorescence est appelée fluorophore. La plupart des échantillons biologiques contiennent des fluorophores endogènes négligeables; par conséquent, les fluorophores exogènes doivent être utilisés pour mettre en évidence les caractéristiques d'intérêt dans l'échantillon. Pendant la microscopie de fluorescence, l'échantillon est éclairé avec la lumière de la longueur d'onde appropriée pour l'absorption par le fluorophore. Lors de l'absorption d'un photon, un fluorophore est dit être «excité» et le processus d'absorption est appelé «excitation». Lorsqu'un fluorophore abandonne l'énergie sous la forme d'un photon, le processus est connu sous le nom d'« émission », et le photon émis est appelé fluorescence.
Le faisceau lumineux utilisé pour exciter un fluorophore est focalisé par la lentille objective d'un microscope et converge à un « point focal » où il est focalement focal. Au-delà du point focal, la lumière diverge à nouveau. Les poutres entrantes et sortantes peuvent être visualisées comme une paire de cônes touchant au point focal (voir la figure 1, panneau gauche). Le phénomène de diffraction impose une limite à la façon dont étroitement un faisceau de lumière peut être concentré - le faisceau se concentre réellement à un endroit de taille finie. Deux facteurs déterminent la taille de la tache focale : 1) la longueur d'onde de la lumière, et 2) la capacité de collecte de lumière de la lentille objective, qui se caractérise par son ouverture numérique (NA). Le «spot» focal s'étend non seulement dans le plan x-y, mais aussi dans la direction z, et est en réalité un volume focal. Les dimensions de ce volume focal définissent la résolution maximale réalisable par l'imagerie optique. Bien que le nombre de photons soit le plus grand dans le volume focal, les chemins de lumière conique au-dessus et au-dessous de la mise au point contiennent également une densité plus faible de photons. N'importe quel fluorophore dans le chemin de lumière peut ainsi être excité. Dans la microscopie conventionnelle (à champ large), les émissions des fluorophores au-dessus et au-dessous du plan focal contribuent à la fluorescence hors foyer (un « fond brumeux »), ce qui réduit la résolution et le contraste de l'image, comme le montre la figure 1, le cube rouge représentant l'émission de fluorophore au-dessus du plan focal (étoile rouge) qui a comme conséquence la fluorescence hors-focus (en haut à droite). Ce problème est amélioré dans la microscopie confocale, en raison de l'utilisation d'un trou d'épingle. (figure 2, en bas à droite). Tel que représenté à la figure 3, le trou d'épingle permet aux émissions provenant du point focal d'atteindre le détecteur (à gauche), tout en empêchant la fluorescence hors foyer (à droite) d'atteindre le détecteur, améliorant ainsi à la fois la résolution et le contraste.

Figure 1. Résolution optique de l'épifluorescence par rapport à la microscopie confocale. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Le faisceau lumineux utilisé pour exciter un fluorophore est focalisé par la lentille objective d'un microscope et converge à un volume focal, puis diverge (à gauche). L'étoile rouge représente le plan focal d'un échantillon qui est représenté tandis que le carré rouge représente l'émission de fluorophore au-dessus du plan focal. Lors de la capture d'une image de cet échantillon à l'aide d'un microscope épifluorescent, l'émission du carré rouge flou sera visible et contribuera à un « fond brumeux » (en haut à droite). Les microscopes confocals ont un trou d'épingle qui empêche la détection de la lumière émise à l'extérieur du plan focal, éliminant le « fond brumeux » (en bas à droite).
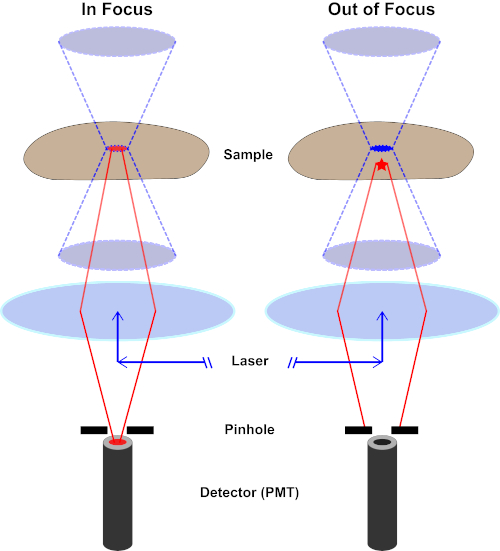
Figure 2. Effet de trou d'épingle dans la microscopie confocale. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Bien que l'intensité la plus élevée de la lumière d'excitation soit au point focal de la lentille (gauche, ovale rouge), d'autres parties de l'échantillon ne se trouve pas dans le point focal (droite, étoile rouge) obtiendront la lumière et la fluoresce. Afin d'éviter que la lumière émise par ces régions floues n'atteigne le détecteur, un écran muni d'un sténopé est présent devant le détecteur. Seule la lumière dans le foyer (à gauche) émise par le plan focal est capable de traverser le sténopé et d'atteindre le détecteur. La lumière hors foyer (à droite) est bloquée avec le sténopé et n'atteint pas le détecteur.

Figure 3. Principaux composants d'un microscope à balayage laser confocal. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Par souci de simplicité, la description mécaniste d'un microscope confocal sera limitée à celle du Nikon Eclipse Ti A1R. Bien qu'il puisse y avoir des différences techniques mineures entre les différents microscopes confocal, l'A1R sert bien comme un bon modèle pour décrire la fonction de microscope confocal. Le faisceau lumineux d'excitation, produit par un tableau de lasers à diodes, est reflété par le miroir dichroïque primaire dans l'objectif, qui concentre la lumière sur le spécimen en cours d'image. Le miroir dichroïque primaire réfléchit sélectivement la lumière d'excitation tout en permettant à la lumière à d'autres longueurs d'onde de passer à travers. La lumière rencontre alors les miroirs de balayage qui balaient le faisceau lumineux à travers le spécimen d'une manière x-y, illuminant un seul (x,y) pixel à la fois. La fluorescence émise par les fluorophores au pixel lumineux est recueillie par l'objectif et passe à travers le miroir dichroïque primaire pour atteindre un tableau de tubes photomultiplicateurs (PMT). Les miroirs dichroiques secondaires dirigent la lumière d'émission vers le PMT approprié. La lumière d'excitation dispersée par l'échantillon dans l'objectif est réfléchie par le miroir dichroïque primaire vers le spécimen, et donc empêchée d'entrer dans la détection voie légère et d'atteindre les PMT (voir la figure 3). Cela permet de quantifier la fluorescence relativement faible sans contamination par la lumière dispersée du faisceau lumineux d'excitation, qui est généralement des ordres de grandeur plus intenses que la fluorescence. Parce que le sténopé bloque la lumière de l'extérieur du volume focal, la lumière arrivant au détecteur provient d'un étroit, sélectionné z-plan. Par conséquent, les images peuvent être recueillies à partir d'une série de z-planesadjacents; cette série d'images est souvent appelée « z-stack ». En utilisant le logiciel approprié, une pile zpeut être traitée pour générer une image 3D du spécimen. Un avantage particulier de la microscopie confocale est la capacité de distinguer la localisation subcellulaire de la coloration. Par exemple, la différenciation entre la coloration de membrane de la coloration intracellulaire, qui est très provocante avec la microscopie conventionnelle d'épifluorescence (1, 2, 3).
La préparation de l'échantillon est une facette importante de l'imagerie confocale. Une force des techniques de microscopie optique est la flexibilité d'imager des cellules vivantes ou fixes. Lorsque vous tentez de produire des images 3D, en raison du nombre d'images qui doivent être acquises pour une z-pile, la difficulté de maintenir la santé cellulaire, et le mouvement des cellules vivantes et de leurs organites, l'utilisation de cellules fixes est typique. La procédure pour fixer et tacher des cellules pour la fluorescence confocale est semblable à celle conventionnellement employée dans l'immunofluorescence. Après la culture dans les diapositives de chambre ou sur les couvertures, les cellules sont fixées à l'aide de paraformaldéhyde pour préserver la morphologie cellulaire. La liaison non spécifique d'anticorps est bloquée utilisant l'albumine bovine de sérum, le lait, ou le sérum normal. Afin de maintenir la spécificité des anticorps secondaires, la solution utilisée ne doit pas provenir de la même espèce dans laquelle les anticorps primaires ont été générés. Les cellules sont incubées avec des anticorps primaires qui lient l'antigène d'intérêt. Lors de l'étiquetage de plusieurs cibles cellulaires, les anticorps primaires doivent être dérivés d'une espèce différente. Les anticorps taguant un antigène sont alors liés par des anticorps secondaires fluorophore-conjugués. Les anticorps secondaires conjugués au fluorophore doivent être sélectionnés afin qu'ils soient compatibles avec les longueurs d'onde de l'excitation laser disponibles dans le microscope confocal. Lors de la visualisation de plusieurs antigènes, les spectres d'excitation/émission des fluorophores devraient différer suffisamment pour que leurs signaux puissent être discriminés par l'analyse microscopique. Le spécimen taché est ensuite monté sur une glissière pour l'imagerie. Un support de montage est utilisé pour prévenir le photoblanchiment et la déshydratation des spécimens. Si vous le souhaitez, un support de montage contenant une contre-tache nucléaire (p. ex. DAPI ou Hoechst) peut être utilisé (4).
Dans le protocole suivant, les fibroblastes de souris transfectés pour exprimer cD1d (LCD1) ont été souillés avec des anticorps reconnaissant CD1d et CD107a (LAMP-1). CD1d est un complexe d'histocompatibilité majeur 1 (MHC 1)-comme le récepteur présent sur la surface des cellules présentant d'antigène qui présente des antigènes. LAMP-1 (protéine de membrane associée lysosomal-1) est une protéine transmembranaire principalement présente dans les membranes lysosomal. Pour la présentation appropriée d'antigène, CD1d est trafiqué par le compartiment lysosomal de pH bas, ainsi LAMP-1 est employé comme marqueur du compartiment lysosomal pour ce protocole. En sondant les cellules LCD1 avec anti-CD1d et anti- LAMP-1 qui ont été produites dans différentes espèces, les anticorps secondaires avec des fluorophores uniques peuvent être utilisés pour déterminer la localisation de chaque protéine dans la cellule et si CD1d est présent dans le LAMP-1 positif compartiments lysosomal.
Procédure
1. Matériaux
Tampons
- Tampon de lavage : 1 X saline stérile tamponnée de phosphate (PBS) sans calcium ni magnésium
- Tampon de fixation : 1% de paraformaldehyde en PBS
- Tampon de perméabilisation : 0,1 % Triton X-100 en PBS
- Tampon de blocage : 1% d'albumine de sérum bovin dans PBS
- Milieu de croissance cellulaire : DMEM complété par 10% de sérum bovin fœtal (FBS), pénicilline/streptomycine, et L-glutamine
équipement
- Hotte d'écoulement laminaire
- Incubateur humidifié (37oC, 5% CO2)
- Microscope à balayage laser confocal; ici, Nikon Eclipse Ti laser
Matériaux et réactifs
- Diapositives de culture cellulaire chambrée
- Médias de montage anti-fade avec DAPI (pour les noyaux de coloration)
- Verre de couverture de microscope
- Lingettes de tâche délicates
- Pipettors et conseils
Réagents spécifiques d'assay
- Cellules adhérentes (cellules primaires ou lignées cellulaires); ici, des fibroblastes de souris transfectés avec CD1d ont été employés (LCD1).
- Anticorps primaires pour détecter les cibles cellulaires; ici, rat anti-souris CD107a (LAMP-1) et souris anti-souris CD1d ont été utilisés.
- Anticorps secondaires conjugués au fluorophore spécifiques aux isotypes d'anticorps primaires; ici anti-rat IgG conjugué à Alexafluor 488 et anti-souris IgG conjugué à Alexafluor 647 ont été utilisés.
2. Protocole
Préparation à la coloration des anticorps
Cellules d'ensemencement
- Resuspendre les cellules d'intérêt dans les médias de croissance.
- Ensuite, ensemencez 500 ll de la suspension cellulaire/par puits dans les puits d'une glissière de chambre à 4 puits. (Ici, les cellules LCD1 ont été ensemiées à 2,5x105 cellules/chambre dans 500 ll de supports de croissance. La densité d'ensemencement peut varier d'une lignée cellulaire à l'autre).
- Incuber la glissière de chambre pendant la nuit dans un incubateur de CO2 de 5 % à 37 oC, pour permettre aux cellules d'adhérer au verre.
- Le lendemain, aspirez les supports de chaque puits, puis lavez les cellules 1X avec 500 L de PBS.
fixation
- Pour fixer les cellules, ajouter une solution de paraformaldéhyde de 500 l 1 % dans chaque puits et incuber pendant 15 min à température ambiante.
- Après l'incubation, recueillir le paraformaldéhyde dans un contenant de déchets liquides dangereux approprié.
- Ensuite, lavez les cellules 3 fois avec 500 L PBS pour enlever les restes du fixatif.
Perméabilisation
- Perméabilisez les cellules en couvant avec un tampon/puits de perméabilisation de 500 l l pendant 15 minutes à température ambiante.
- Ensuite, lavez les cellules brièvement 3 fois avec 500 L de PBS.
Blocage
- Incuber les cellules dans chaque puits avec un tampon de blocage de 500 l pendant 1 heure à 4 oC, pour bloquer la liaison non spécifique des anticorps.
Incubation primaire d'anticorps
- Aspirez le tampon de blocage des chambres de diapositives.
- Ensuite, ajoutez 500 L de solution d'anticorps primaire diluée aux cellules. (Ici, anti-CD107a (LAMP-1) a été dilué 1:500 et anti-CD1d a été utilisé non dilué (1H6 anticorps monoclonaux a été gentiment fourni par le Dr Randy Brutkiewicz)).
- Incuber les toboggans pendant la nuit à 4oC.
Remarque : Si vous probez pour plus d'une cible, assurez-vous que les anticorps primaires sont différents isotypes. Les concentrations d'anticorps recommandées varient d'un fabricant à l'autre et doivent être titrées avant d'être utilisées.
Incubation secondaire d'anticorps
- Aspirez la solution d'anticorps primaire des puits.
- Laver les chambres de puits 4 fois avec 500 L PBS.
- Ensuite, ajoutez 500 L de la solution d'anticorps secondaire diluée à chaque puits. (Ici, les anticorps secondaires- anti-souris IgG Alexafluor 647 et anti-rat IgG Alexafluor 488 ont été dilués 1:2000 dans le tampon de blocage).
- Incuber à température ambiante pendant 1 h dans l'obscurité.
- Après l'incubation, aspirez la solution d'anticorps secondaire.
- Laver les chambres 4 fois avec 500 L PBS pour enlever tout anticorps secondaire non lié.
Remarque : Les concentrations d'anticorps recommandées varient d'un fabricant à l'autre et doivent être titrées avant d'être utilisées. Si l'on cherche plus d'une cible, les anticorps secondaires doivent être conjugués à différents fluorophores avec des spectres d'excitation/émission uniques. Gardez également à l'esprit l'excitation/émission de la contretache nucléaire (c.-à-d. DAPI) tout en sélectionnant les fluorophores. La sélection de fluorophore peut être affectée par la configuration laser du microscope confocal utilisé. La configuration laser de la machine dictera quels fluorophores sont appropriés pour l'expérience.
3. Les couvertures de montage
- Tout d'abord, retirez soigneusement les chambres de la glissière.
- Ensuite, maintenez la glissière à l'angle au-dessus d'une lingette délicate de tâche et retirez le fluide des bords sans toucher les cellules.
- Ajouter 1 goutte de milieu de montage antifade, contenant la tache nucléaire DAPI, sur chaque section de cellules.
- Ensuite, placez un bordereau de 20 mm x 60 mm sur la glissière en la tenant sur les bords à l'aide du bout des doigts. (Éviter la formation de bulles sur les cellules, car elles interfèrent avec l'imagerie).
- Essuyez tout support de montage supplémentaire sur les côtés avec une manette délicate et rangez les diapositives dans l'obscurité à température ambiante jusqu'à une semaine.
4. Imagerie confocale
Échantillons d'image sur un microscope à balayage laser confocal. Pour les données présentées dans la figure 2, le Nikon Eclipse Ti A1R a été utilisé avec le logiciel NIS Elements Advanced Research. La section suivante détaille la procédure de capture d'images à l'aide du logiciel susmentionné.
- Pour commencer l'imagerie des cellules, ouvrez le logiciel de recherche avancéeNIS Elementsen cliquant sur l'icônelogicielle NIS.
- Ensuite, sur la fenêtre de contrôle cliquez sur 'TiPad' onglet et choisissez l'objectif souhaité pour l'imagerie. (Ici, le premier objectif 40x a été utilisé).
- Chargez la diapositive avec des cellules sur la scène et centrez-la sous la lentille.
- Maintenant, sur l'onglet 'A1plus Compact GUI',configurez les lasers appropriés pour les fluorophores utilisés. Cliquez sur le symbole de l'engrenage pour ouvrir le menu de teinture et de réglages spectrals et sélectionnez les canaux nécessaires et définir le laser pour chaque canal.
- Ensuite, sélectionnez les émissions appropriées dans le menu déroulant sous le premier miroir dichroïque.
- Ensuite, sous 'A1plus Compact GUI' fenêtre, cliquez sur 'Ch. Series' pour mettre en place la série de canaux de ligne, qui met en place si les lasers utilisés tirera sur l'échantillon simultanément ou séquentiellement. (Ici, les laissez-passer séquentiels ont été choisis, à commencer par le canal 1, suivi par le canal 2, puis 4).
- Après cela, commencez à numériser en cliquant sur l'icône 'Arrow tip'sur le dessus. À ce stade, alors que l'imagerie est en direct, sous 'A1plus Compact GUI' fenêtre, cliquez sur l'échelle coulissante et modifier la taille du sténopé pour assurer la limitation de la lumière de mise au point. (Ici, le réglage le plus bas disponible (0,5), a été utilisé).
- Ensuite, ajustez les paramètres «haute tension» et «offset» sous chaque laser à des niveaux appropriés, en utilisant les échelles coulissantes pour permettre la détection de la coloration spécifique tout en limitant toute coloration de fond potentielle. Si un échantillon de coloration positive est disponible, commencez par l'imagerie de cet échantillon pour chaque canal afin de s'assurer que les réglages laser donnent des rapports signaux/bruit optimaux.
Attention : une intensité laser élevée pendant de longues périodes peut provoquer le photoblanchiment. - Après avoir défini les valeurs HV et offset optimales pour chaque laser, cliquez sur l'onglet 'ND Acquisition'et sélectionnez ensuite l'icône 'Z'pour configurer les paramètres de la série Z. Ensuite, lors de l'acquisition d'une image en direct de l'échantillon, d'abord définir le fond en trouvant le bas de l'image et en cliquant sur le bouton«bas»puis trouver la position supérieure de l'échantillon et cliquez sur le bouton'haut'. Définir la taille de l'étape soit en tapant spécifiquement la taille d'étape préférée en m pour chaque étape ou en spécifiant le nombre total d'étapes nécessaires.
- Une fois que les paramètres de la série Z ont été réglés, sélectionnez la résolution de taille/pixel désirée de l'image. Pour ce faire, cliquez sur la fenêtre 'A1plus Compact GUI' et sous l'icône 'taille' sélectionnez la résolution désirée. Pour diminuer le bruit de l'image, on peut sélectionner le menu déroulant à côté du symbole'' ' pour faire la moyenne du nombre d'images sélectionnées.
- Maintenant, cliquez sur l'onglet 'Run now' sur le menu 'ND Acquisition' afin de commencer à imagerie de l'échantillon.
- Une fois l'imagerie terminée, enregistrez l'image en cliquant sur 'File', puis 'Save As', qui exportera le fichier d'image avec l'extension'.nd2'. Enfin, répétez le processus pour chacun des autres échantillons.
Résultats
Dans cette expérience, les fibroblastes de souris exprimant le gène de glycoprotéine de surface CD1d ont été fixés, immunostained et imaged sur un microscope confocal. Une image représentative obtenue à l'aide du protocole ci-dessus est indiquée à la figure 4. Dans le panneau supérieur de A, des images monocanaux montrant le motif de coloration de chaque cible individuelle sont présentées. Ces images comprennent une seule section (tranche) de la z-stack capturée. Le panneau droit montre la coloration DAPI des noyaux des cellules. Les panneaux du centre montrent CD1d taché en rouge et LAMP-1, un marqueur lysosomal, taché en vert. Le panneau de gauche est une image composite où les trois canaux différents sont fusionnés. L'apparition de résultats jaunes du chevauchement des canaux rouges et verts, et indique une zone où CD1d et LAMP-1 sont co-localisés. Les résultats de la coloration confirment que le CD1d est localisé dans les compartiments endosomal LAMP-1MD. Il ya aussi des zones où une seule couleur est présente, ce qui indique la présence de CD1d ou LAMP-1 sans co-localisation. Le panneau inférieur de A montre un rendu 3D des cellules construites à partir d'images capturées dans la z-pile.
Le panneau B montre une tranche de la z-pile à 100x grossissement démontrant les modèles d'expression de ces deux protéines plus en détail. La boîte rose sur le côté droit de l'image affiche la section transversale de la x-coordinate désignée par la ligne rose dans l'image, qui représente la vue latérale à la ligne rose. De même, la boîte bleue sur le bas de l'image montre la section transversale de la y-coordinate désignée par la ligne bleue dans l'image, qui représente la vue avant à la ligne bleue. Le rendu 3D de l'image z-stack permet aux utilisateurs de visualiser l'image en 3D, en visualisant tous les plans x, y et z.
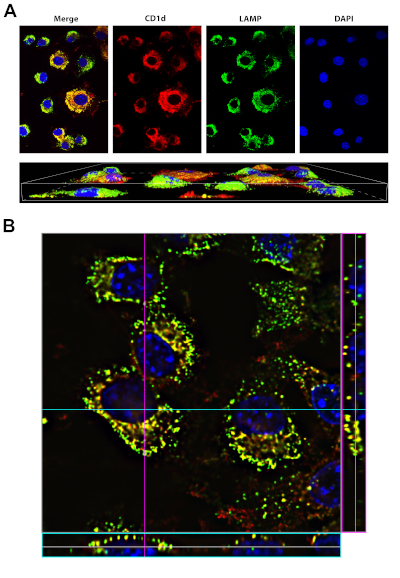
Figure 4 : Coloration de CD1D et LAMP1. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
A, panneau supérieur: Les cellules LCD1 ont été fixées, perméabilisées et tachées d'anticorps à CD1d (rouge) et LAMP-1 (vert, un marqueur du compartiment lysosomal). DAPI (bleu, a été utilisé pour visualiser le noyau). La fusion (panneau gauche) montre que le CD1d est localisé dans le compartiment endosomal tardif/lysosomal positif de LAMP-1 (jaune).
A, panneau inférieur : rendu 3D des mêmes cellules dans le panneau supérieur. Les images ont été acquises à l'aide d'un objectif d'immersion d'huile 40x sur le Nikon Eclipse Ti, en utilisant le logiciel NIS Elements Advanced Research.
B: 100x image des cellules LCD1d tachées comme en A, avec des informations de pile pour un y-coordinate particulier (dénoté par la ligne bleue) sur le bas de l'image (boîte bleue). Les informations de pile pour une x-coordinate particulière (dénotée par la ligne rose) sont affichées sur le côté droit de l'image (boîte rose).
Applications et Résumé
La coloration fluorescente confocale est une procédure relativement simple qui donne lieu à des images de très haute qualité de spécimens qui sont préparés de la même manière que pour la microscopie à fluorescence conventionnelle. En bref, les échantillons sont fixes, perméabilisés, puis bloqués. Les anticorps primaires contre une protéine ou des protéines d'intérêt sont autorisés à se lier, puis des anticorps secondaires conjugués au fluorophore sont utilisés pour visualiser la coloration. La microscopie par fluorescence confocale a des applications dans de nombreux domaines de recherche. Par exemple, en taclant des marqueurs d'organites sous-cellulaires avec une protéine d'intérêt, la microscopie confocale peut être utilisée pour déterminer les emplacements subcellulaires de diverses protéines. Par rapport à la microscopie fluorescence conventionnelle, l'imagerie confocale peut mieux distinguer entre la surface cellulaire et l'emplacement intracellulaire d'une protéine. En outre, la formation image confocale peut également être utilisée pour déterminer si deux protéines co-localisent dans la cellule. Bien qu'elle ne soit pas décrite dans ce protocole, la microscopie confocale de fluorescence peut également être effectuée sur des cellules vivantes pour détecter des changements dynamiques.

Vidéo 1: Vidéo créée dans le logiciel NIS Elements Advanced Research, mettant en évidence la capacité de se déplacer à travers le rendu 3D des images. S'il vous plaît cliquez ici pour voir cette vidéo (Cliquez à droite pour télécharger).
Tags
Passer à...
Vidéos de cette collection:

Now Playing
Microscopie confocale à fluorescence : une technique pour localiser les protéines dans les fibroblastes de souris
Immunology
43.4K Vues

Cytométrie en flux et tri cellulaire activé par fluorescence (FACS) : isolement des lymphocytes B spléniques
Immunology
93.2K Vues

Tri cellulaire magnétique (MACS) : isolement des lymphocytes T thymiques
Immunology
23.1K Vues

Tests ELISA : Indirect, en sandwich et par compétition
Immunology
239.4K Vues

Test ELISPOT : Détection des splénocytes sécrétants l'IFNgamma
Immunology
28.8K Vues

Immunohistochimie et immunocytochimie : Imagerie tissulaire par microscopie optique
Immunology
79.2K Vues

Génération d'anticorps monoclonaux à l'aide d'hybridomes
Immunology
43.7K Vues

Microscopie à fluorescence : coloration par immunofluorescence des sections de tissus inclus en paraffine
Immunology
54.0K Vues

Techniques basées sur l'immunoprécipitation : purification des protéines endogènes à l'aide de billes d'agarose
Immunology
87.9K Vues

Analyse du cycle cellulaire : utilisation de la coloration CFSE et de la cytométrie de flux pour évaluer la prolifération des lymphocytes T CD4 et CD8 après stimulation
Immunology
24.3K Vues

Transfert adoptif de cellules : introduction de splénocytes d'une souris donneuse vers une souris hôte et évaluation du taux de succès au FACS
Immunology
22.6K Vues

Test de mort cellulaire : libération du chromium pour mesurer la cytotoxicité
Immunology
151.5K Vues
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.