
Method Article
Un método rápido y cuantitativo para la variante y modificación poste-de translación activado mapeo de péptidos genomas
En este artículo
Resumen
Aquí presentamos la herramienta proteogenomic PoGo y protocolos para la modificación rápida, cuantitativo, poste-de translación y variante activado mapeo de péptidos identificados a través de espectrometría de masas en los genomas de referencia. Esta herramienta es de uso para integrar y visualizar proteogenomic y estudios proteómicos personal con datos de genómica ortogonal.
Resumen
La diafonía entre los genes, transcritos y proteínas es la clave de respuestas celulares; por lo tanto, el análisis de niveles moleculares como entidades distintas lentamente se extiende a estudios integrativos para mejorar la comprensión de la dinámica molecular dentro de las células. Herramientas actuales para la visualización y la integración de la proteómica con otros conjuntos de datos ómicos son inadecuadas para estudios a gran escala. Además, capturan sólo secuencia básica identificar, descartar modificaciones post-traduccionales y cuantificación. Para enfrentar estos problemas, hemos desarrollado PoGo para péptidos con modificaciones post-traduccionales asociadas y cuantificación para anotación del genoma de referencia. Además, la herramienta fue desarrollada para permitir el mapeo de péptidos identificados de bases de datos de secuencia personalizada incorporando variantes solo aminoácido. Mientras que el PoGo es una herramienta de línea de comandos, la interfaz gráfica PoGoGUI permite a los investigadores de la bioinformática no fácilmente mapa péptidos a 25 especies apoyadas por anotación del genoma de Ensembl. La salida generada toma los formatos de archivo en el campo de la genómica y, por lo tanto, la visualización es compatible en la mayoría de los navegadores de genoma. Para estudios a gran escala, PoGo es apoyada por TrackHubGenerator para crear repositorios web accesible de datos asignados a genomas que también permiten un fácil intercambio de datos de proteogenomics. Con poco esfuerzo, esta herramienta puede asignar millones de péptidos a genomas de referencia dentro de pocos minutos, superando a otras herramientas disponibles secuencia-identidad basada. Este protocolo muestra los mejores enfoques para la asignación de proteogenomics a través de PoGo con conjuntos de datos públicamente disponibles de cuantitativa y fosfoproteómico, así como estudios de gran escala.
Introducción
En las células, genoma, transcriptoma y proteoma afectan para modular una respuesta a los estímulos internos y externos e interactuar con otros para llevar a cabo funciones específicas hacia la salud y la enfermedad. Por lo tanto, caracterizar y cuantificar genes, transcritos y proteínas es crucial para comprender cabalmente los procesos celulares. Secuenciación de próxima generación (NGS) es una de las estrategias más comúnmente aplicadas para identificar y cuantificar la expresión génica y la transcripción. Sin embargo, expresión de la proteína es comúnmente evaluada por espectrometría de masas (MS). Avances significativos en tecnología MS durante la última década ha permitido más una completa identificación y cuantificación de proteomas, hacer los datos comparables con transcriptómica1. Proteogenomics y multi-ómicas como formas de integrar datos NGS y MS se han convertido en poderosos enfoques para evaluar procesos celulares a través de múltiples niveles moleculares, identificar subtipos de cáncer y conduce a nuevos objetivos potenciales de la droga en cáncer2 , 3. es importante tener en cuenta que proteogenomics fue utilizado inicialmente para proporcionar evidencia de la proteómica para gene y transcripción de las anotaciones4. Varios genes se pensaba que no codificante recientemente han sido sometidos a reevaluación considerando tejido humano a gran escala datos5,6,7. Además, los datos proteómicos se utilizan con éxito para apoyar los esfuerzos de anotación en organismos no-modelo8,9. Sin embargo, proteogenomic integración de datos pueden ser aprovechados además de resaltar la expresión de proteína en relación a características genómicas y dilucidar entre transcripciones y proteínas proporcionando un sistema de referencia combinado y métodos para visualización conjunta.
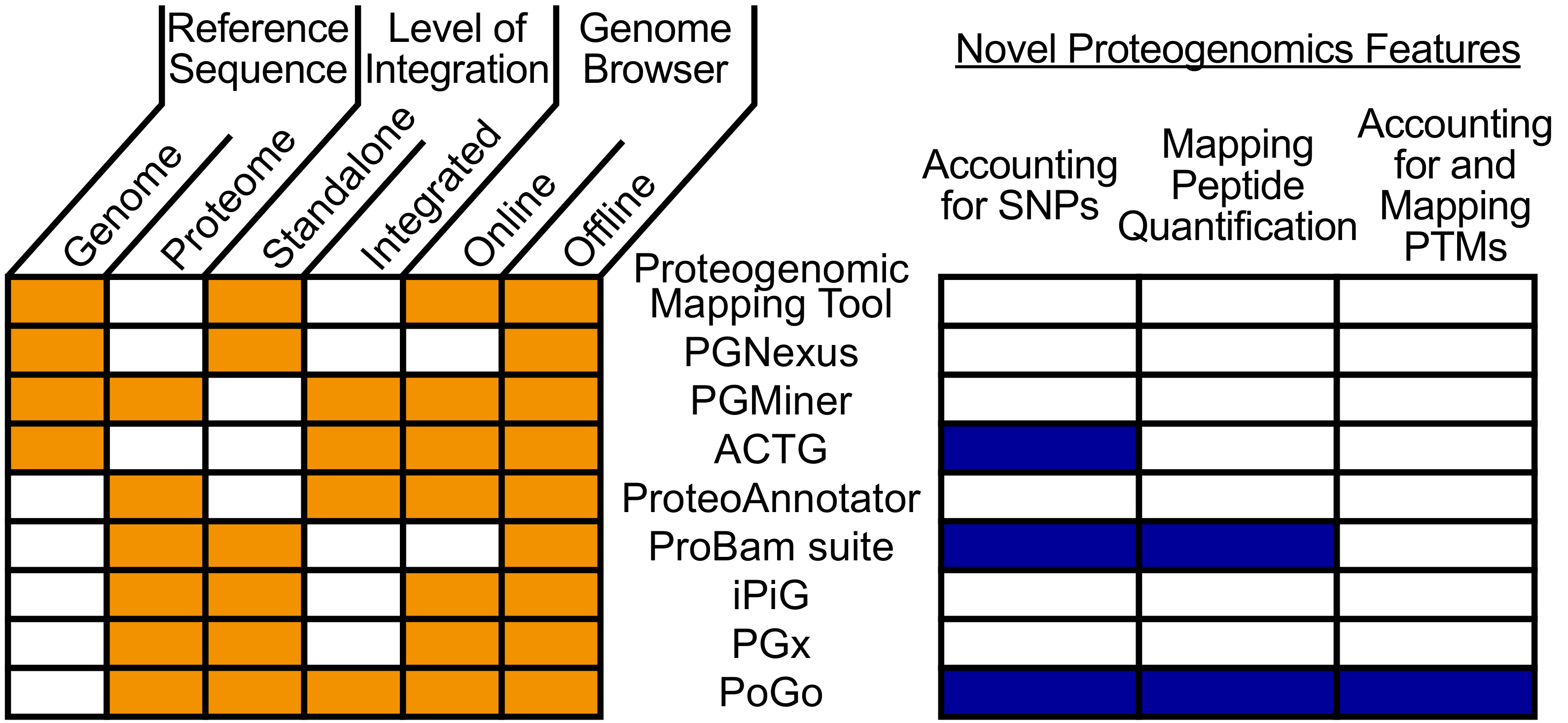
Con el fin de proporcionar una referencia común para datos de genómica, transcriptómica y proteómica, se han implementado numerosas herramientas para péptidos de mapeo identificadas a través de MS sobre genoma coordenadas10,11,12 ,13,14,15,16,17. Enfoques difieren en aspectos tales como la referencia de la cartografía, soporte de navegadores de genoma y el grado de integración con otras herramientas de proteómica como se muestra en la figura 1. Mientras que algunas herramientas mapa de péptidos traducción inversas en un genoma16, otros utilizan una posición de búsqueda motor anotado en una anotación de la proteína y gen para reconstruir la secuencia de nucleótidos del péptido15. Todavía otros utilizan una traducción de 3 o 6 marco del genoma a péptidos contra11,13. Por último, varias herramientas saltar las secuencias de nucleótido y utilizan las traducciones de secuencia del aminoácido de transcritos de RNA-secuencia asignada como intermedio para asignar péptidos a genoma asociado coordenadas10,12, 14,17. Sin embargo, la traducción de secuencias de nucleótidos es un proceso lento y bases de datos personalizadas son propensos a errores que se propagan a la asignación de péptido. Para el mapeo rápido y alto rendimiento, una referencia pequeña y completa es fundamental. Por lo tanto, una referencia estandarizada proteína con coordenadas genoma asociado es esencial para péptido precisa cartografía del genoma. Aspectos novedosos en proteogenomics, como la incorporación de variantes y modificaciones post-traduccionales (PTMs)2,3, están ganando impulso a través de estudios recientes. Sin embargo, estos generalmente no son compatibles con proteogenomic actual asignación de herramientas como se muestra en la figura 1. Para mejorar la velocidad y la calidad de la cartografía, PoGo se desarrolló una herramienta que permite la asignación rápida y cuantitativa de los péptidos a genomas18. Además, PoGo permite el mapeo de péptidos con hasta dos variantes y modificaciones postraduccionales anotadas.
PoGo se ha desarrollado para enfrentar el rápido aumento de cuantitativa conjuntos de datos de alta resolución captura de proteomas y modificaciones globales y proporciona una herramienta central para los análisis a gran escala como variación personal y medicina de precisión. Este artículo describe el uso de esta herramienta para visualizar la presencia de modificaciones post-traduccionales en lo referente a características genómicas. Además, este artículo destaca la identificación de eventos alternativos que empalma a través de péptidos asignadas y el mapeo de péptidos identificados a través de bases de datos variante personalizados para un genoma de referencia. Este protocolo utiliza conjuntos de datos públicamente disponibles de orgullo archivo19 al demostrar estas funcionalidades de PoGo. Además, este protocolo describe el uso de TrackHubGenerator para la creación de centros en línea accesibles de péptidos mapeados genomas para estudios a gran escala proteogenomics.
Protocolo
1. preparación, descarga y configuración
Nota: Los ejemplos de ruta de archivo y la carpeta se muestran en un formato de Windows por la facilidad de acceso para los usuarios estándar. PoGo y PoGoGUI también están disponibles para sistemas de operativos Linux y macOS.
-
Descargar PoGo y PoGoGUI de GitHub
- Abra un navegador web y vaya a PoGo en GitHub (http://github.com/cschlaffner/PoGo/). Seleccione Releases y descargar el última versión zip archivo comprimido. Extraiga el archivo comprimido en la carpeta de ejecutables (por ejemplo, C:\PoGo\executables\).
- Navegar en el navegador web para PoGoGUI en GitHub (http://github.com/cschlaffner/PoGoGUI/). Seleccione Releases y descargar el última versión archivo jar (por ejemplo, "PoGoGUI-v1.0.2.jar"). Guarde el archivo jar en la carpeta de ejecutables.
-
Descargar la anotación del genoma y secuencias de codificación de la proteína traducidas
Nota: Bajar la anotación del genoma y secuencias de codificación de la proteína traducidas para especies compatibles de GENCODE7 (www.gencodegenes.org) o Ensembl20 (www.ensembl.org) en el formato de transferencia General (GTF) y las secuencias de la proteína en Formato FASTA.- En el explorador web, desplácese hasta www.gencodegenes.org y seleccione datos | Humano | Versión actual. La anotación completa del gen mediante el enlace GTF de descargar y extraer el archivo gz comprimido en la carpeta de datos (por ejemplo, C:\PoGo\Data\) utilizando un programa de descompresión (por ejemplo, 7-Zip).
- Descargar las secuencias de traducción transcripción proteína de codificación a través del enlace FASTA y extraiga el archivo gz comprimido en la carpeta de datos generada en el paso anterior.
- También puede navegar en el navegador web para www.ensembl.org y seleccione descargas seguido por descarga de datos vía FTP. Encontrar una especie de soporte (e.g., humanos). Descargar el último archivo de liberación para la anotación de transcripción mediante el enlace de la GTF en la columna de conjunto de genes . Elija el archivo con la estructura de nombre "species.release.gtf.gz" y extraer el archivo gz comprimido en la carpeta data.
- Descargar la última versión de codificación de proteínas transcripción traducción secuencias usando la FASTA enlace en la columna de la secuencia de la proteína (FASTA) . Elija el archivo con la estructura de nombre "species.release.pep.all.fa.gz" y extraer el archivo gz comprimido en la carpeta data.
-
Preparar péptido archivos de identificación
Nota: PoGo sólo admite un formato de 4 columnas que contiene identificador muestra, secuencia de péptido, cantidad de péptido-espectro-partidos (PSMs) y valor cuantitativo. Sin embargo, PoGoGUI soportes de identificación estandarizados archivo formatos mzIdentML, mzid y mzTab y convierte en formato de 4 columnas de PoGo con el marco disponible para el público api de la base de la datos de la ms21. Archivos en formato mzTab, mzid o mzIdentML pueden descargarse del orgullo archivo19. Como alternativa, pueden proporcionar datos en un formato de archivo de ficha separado con la extensión .tsv o .pogo. El formato contiene 4 columnas con los encabezados de columna siguientes: identificador de la muestra (muestra), secuencias del peptide (péptido), número de péptido-espectro-partidos (PSMs) y la cuantificación del péptido (Quant). En la figura 2se muestra un ejemplo.- Descargar un archivo de ejemplo en formato de mzTab de un estudio de proteómica en testículo humano del orgullo archivo19 (https://www.ebi.ac.uk/pride/archive/projects/PXD006465/files22).
- Guardar y extraer el archivo gz comprimido en la carpeta de datos creada en el punto 1.2.1.
Nota: Alternativamente, descargar los datos de ejemplo para fosfoproteómico humano buscado con MaxQuant desde el archivo de orgullo (archivo "Traktman_2013_MaxQuantOutput-completo.zip" del https://www.ebi.ac.uk/pride/archive/projects/PXD005246/files23). - Guardar y extraer el archivo comprimido zip en la carpeta de datos que creó en el paso 1.2.1.
- Abrir una hoja de cálculo en blanco, importar el archivo peptides.txt desde la carpeta C:/PoGo/datos/Traktman_2013_MaxQuantOutput-completo/combinado/txt y mediante la opción datos | De texto/CSV. En la ventana que se abre, haga clic en Editar.
- Eliminar todas las columnas con la excepción de "Secuencia", "Experimento BR1", "Experimento BR2", "Experimento BR3", "Relación H/L normalizó BR1", "Relación H/L normalizó BR2" y "Relación H/L normalizó BR3".
- Seleccione las columnas "Relación H/L normalizó BR1", "Relación H/L normalizó BR2" y "Relación H/L normalizó BR3" y haga clic en transformación | UnPivot columnas. Seleccione las columnas "Experimento BR1", "Experimento BR2" y "Experimento BR3" y repetir la operación unpivot.
- Seleccione la columna resultante "Atributo" y dividir el contenido utilizando transformación | Fractura de columna | Por delimitador. Seleccione espacio como delimitador en el menú desplegable. Repetir la operación de columna "Attribute.1".
- Quitar las columnas resultantes "Attribute.1.1", "Attribute.2", "Attribute.3" y "Attribute.1.1.1".
- Agregar una columna mediante el Agregar columna | Columna personalizada opción. Adaptar la fórmula de columna personalizado para representar lo siguiente: «= [Attribute.4]=[Attribute.1.2]».
- Aplicar un filtro a la columna personalizada generada para filtrar todas las líneas que contengan "FALSE"; serán siendo sólo las líneas que contiene "TRUE".
- Quitar las columnas "Attribute.1.2" y "Custom" y cambiar el orden de las columnas restantes lo siguiente: "Attribute.4", "Secuencia", "Value.1" y "Valor".
- Cambiar los nombres de columna para el "Experimento", "Péptido", "PSMs" y "Quant", respectivamente. Cargar el archivo con Inicio | Cerrar y cargar.
- Guarde el archivo como un archivo delimitado por tabuladores con archivo | Guardar como y seleccione el tipo "Texto (delimitado por tabuladores) (*.txt)". Cambiar el nombre a "peptides_pogo.txt" y guárdelo en la carpeta C:/PoGo/datos.
2. mapeo de péptidos con modificaciones post-traduccionales anotadas y la visualización incluyendo cuantificación
Nota: El archivo de salida resultante se puede cargar en cualquier navegador de genoma que soporte el formato de datos Extensible navegador (cama). Una selección de los navegadores es el navegador de genoma integrante (IGV)24 (que se utiliza en el siguiente), el Browser del genoma de UCSC25y el Browser del genoma de Ensembl20. Es importante tener en cuenta que la anotación GMT proteína FASTA versiones y usadas para cartografiar la PoGo coincida con la versión del genoma en el browser del genoma. Para comunicados de Ensembl humanos 57-75 y GENCODE versiones 3d-19, usar GRCh37/hg19; para las versiones de Ensembl 76 o superiores y GENCODE 20 o superior, utilice GRCh38/hg38. Para las versiones de Ensembl de ratón 74 o superiores y GENCODE M2 o superior, utilice GRCm38.
-
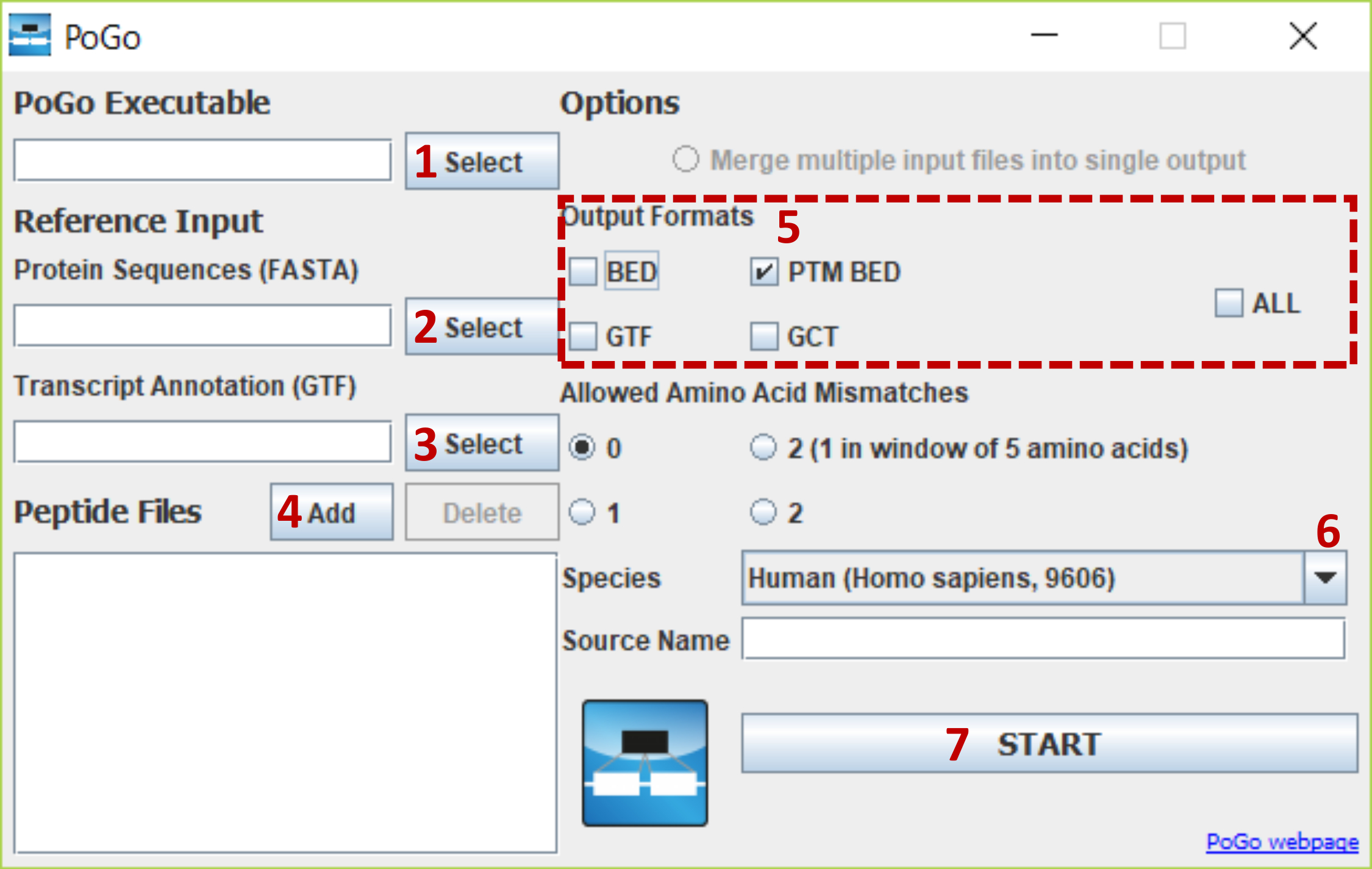
Mapa de péptidos usando PoGoGUI (ver figura 3).
- Desplácese a la carpeta de ejecutables. Inicie el programa haciendo doble clic en el icono de PoGoGUI-vX.X.X.jar.
Nota: La interfaz de usuario gráfica puesta en marcha y permiten la fácil y visual selección de opciones. - Utilice los botones de al lado del ejecutable de"PoGo" . Luego, navegar en la carpeta de ejecutables en la subcarpeta de sistemas operativos relevantes (por ejemplo, C:\PoGo\Executables\Windows\). Seleccionamos el ejecutable de PoGo (por ejemplo, PoGo.exe) y confirme la selección haciendo clic en el botón abrir .
- Seleccione el archivo de entrada de referencia de secuencias de la proteína haciendo clic en seleccionar. Vaya a la carpeta de datos y seleccione el archivo de traducción de FASTA. Confirme la selección haciendo clic en el botón abrir .
- Seleccione el archivo de anotaciones de la transcripción mediante el botón seleccionar . Desplácese a la carpeta de datos y seleccione el archivo GTF de anotación. Confirme la selección haciendo clic en el botón abrir .
- Agregar el archivo de identificación de péptidos, selección múltiple de archivos está habilitada, mediante el botón Agregar al lado de "Archivos de péptidos". Seleccione un archivo en formato compatible mzTab, mzIdentML o mzid, o en el formato de 4 columnas separados por tab descargado y preparado en el paso 1.3.
- Desmarque las casillas de verificación junto a la cama y GTF en la selección de formatos de salida. Sólo salir de cama de PTM y GCT comprobado.
- Seleccionar las especies apropiadas para los datos de la selección de lista desplegable. Es esencial que el archivo FASTA, el archivo GTF y la selección de la lista desplegable son de la misma especie.
- Inicio mapas haciendo clic en el botón Inicio .
Nota: Si es necesario, PoGoGUI será convertir el archivo de entrada en formato de pogo, proporcionar los archivos de pogo en la misma carpeta para mayor comodidad futuro y comenzar el proceso de asignación. La conversión de un archivo de solo mzTab descargado en el paso 1.3.1 durará entre 10-20 minutos antes de empezar la asignación.
- Desplácese a la carpeta de ejecutables. Inicie el programa haciendo doble clic en el icono de PoGoGUI-vX.X.X.jar.
-
Visualizar en el visor de genómica Integrativa
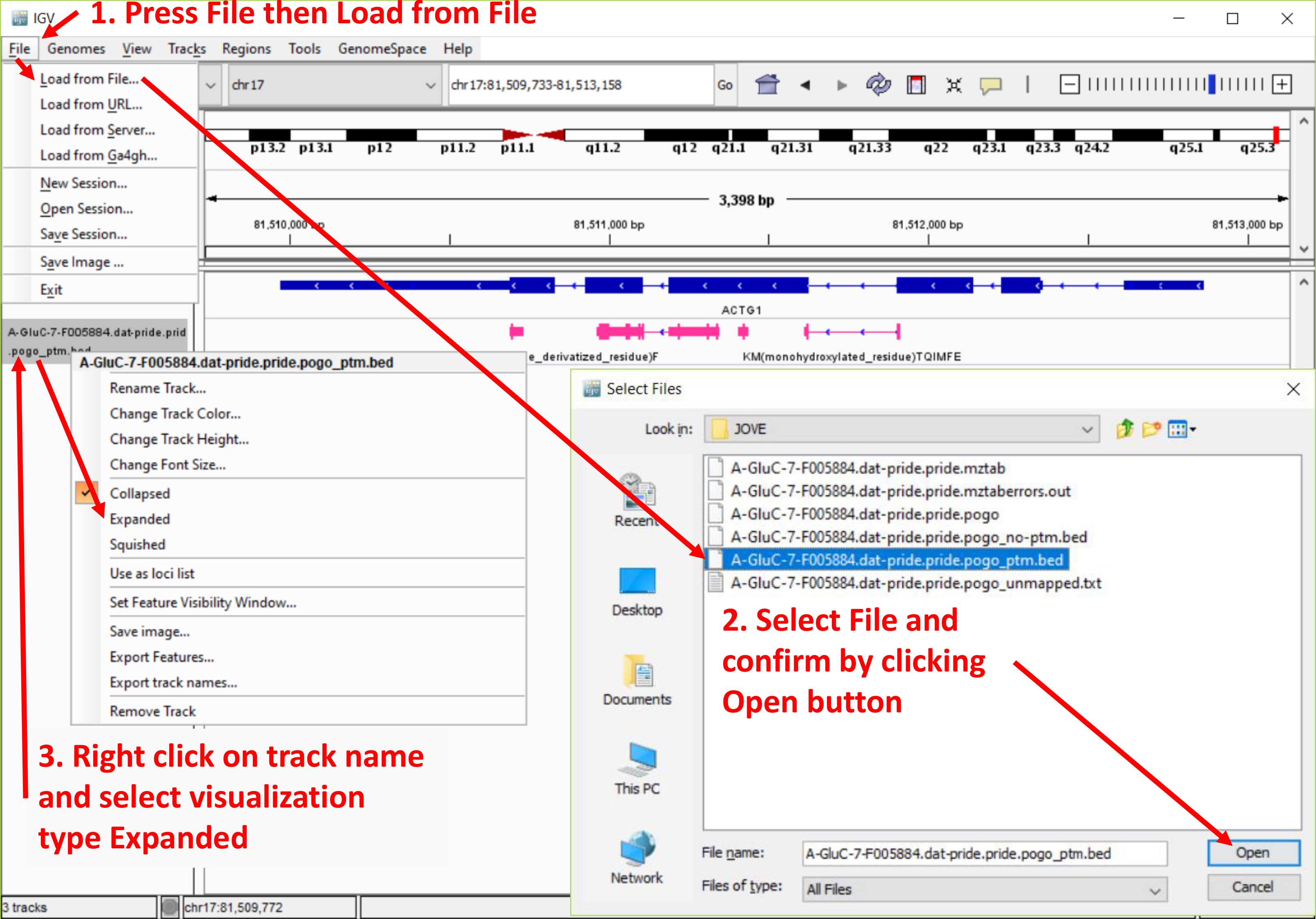
Nota: Consulte la figura 4.- Cargar el archivo de salida de PoGo terminando en "_ptm.bed" en el IGV a través de archivo | Carga de archivo y seleccione el archivo.
Nota: Debido al tamaño, algunos archivos pueden requerir la generación de un índice para permitir una recarga rápida de las regiones genómicas. El IGV pide al usuario automáticamente a la generación. Siga las instrucciones indicadas. - Repita el paso de carga para el archivo termina en "_noptm.bed". Este archivo contiene los péptidos encontrados sin ninguna modificación.
- Tenga en cuenta que cada archivo cargado aparecerá como pistas separadas con el nombre de archivo identifica la pista. Reordenar las pistas arrastrándolos y colocándolos en la posición deseada en la lista.
- Tenga en cuenta que cada pista se muestra inicialmente en la forma contraída. Para ampliarlas, haga clic derecho sobre el nombre de la pista y seleccione expandido para una vista completa de los péptidos incluyendo las secuencias o aplastado para una vista apilada.
- Repita el paso de carga para el archivo termina en ".gct". Este archivo contiene la cuantificación del péptido por ejemplo anotado.
- A diferencia de los archivos cargados, se cargará cada muestra anotado como una pista separada. Reorganizar las muestras a través de arrastrar y soltar las operaciones.
- Navegar dentro del genoma mediante la selección de un cromosoma en el menú desplegable, escriba coordenadas genómicas, buscar un símbolo del gene, o haga clic en y mantenga presionado para seleccionar una sección de un cromosoma para acercar.
- Cargar el archivo de salida de PoGo terminando en "_ptm.bed" en el IGV a través de archivo | Carga de archivo y seleccione el archivo.
3. mapeo de péptidos identificados a través de una base de datos personalizada variante a un genoma de referencia
Nota: PoGo asignación posible usando la interfaz gráfica de usuario (GUI) o a través de la interfaz de línea de comandos. Son intercambiables. En esta parte del Protocolo, la interfaz de línea de comando se utiliza para resaltar la capacidad de intercambio. La segunda parte de esta sección del protocolo requiere que el software herramienta R26. Asegúrese de que está instalado el paquete.
- Mapa de los péptidos de referencia al genoma de referencia.
- Abra un símbolo del sistema (cmd) y desplácese a la carpeta de ejecutables de PoGo (por ejemplo, C:\PoGo\Executables\).
- Escriba el comando siguiente:
PoGo.exe - gtf \PATH\TO\GTF - fasta \PATH\TO\FASTA-en \PATH\TO\IN-formato de cama-especies MYSPECIES- Sustituir el \PATH\TO\GTF, \PATH\TO\FASTA y \PATH\TO\IN con senderos y la anotación GTF, secuencia de la proteína FASTA, archivo de identificación de péptidos (en el formato de 4 columnas con archivo ".tsv" o ".pogo") respectivamente. También sustituir la MYSPECIES con la especie consistente con los datos (por ejemplo, humanos).
- Confirmar la ejecución pulsando la tecla "Enter". Espere hasta que la ejecución haya terminado antes de progresar cualquier aún más.
Nota: Esto puede tardar unos minutos. El fichero resultante se almacenará en la misma carpeta que el archivo de entrada del péptido y se considerará como \PATH\TO\OUT.pogo.bed en el siguiente.
- Extraer sólo variante péptidos desde el archivo de entrada.
- Abierta R y la entrada de carga de archivo \PATH\TO\IN utilizando el siguiente comando:
datosEntrada <-read.table("PATH/TO/IN",header=TRUE,sep="\t") - Carga los péptidos ya asignados mediante el comando:
mappedpeptides <-read.table("PATH/TO/OUT.pogo.bed",sep="\t",header=FALSE) - Eliminar los péptidos que ya fueron asignados de la datosEntrada:
peptidesnotmapped <-datosEntrada [! () datosEntrada$ péptido % en mappedpeptides % $V4)] - Imprimir los péptidos no asignados en un nuevo archivo de entrada:
Write.Table (peptidesnotmapped, "PATH\TO\IN.notmapped.pogo", header = FALSE, sep = "\t", col.names=TRUE,row.names=FALSE,quote=FALSE)
- Abierta R y la entrada de carga de archivo \PATH\TO\IN utilizando el siguiente comando:
- Mapa de los péptidos restantes al genoma de referencia que permite las uniones mal hechas.
- Como en el paso 3.1, abra el símbolo del sistema y desplácese a la carpeta de ejecutables de PoGo.
- Escriba el comando abajo permitiendo desajuste del aminoácido 1 y sustituir el \PATH\TO\GTF, \PATH\TO\FASTA y \PATH\TO\IN.notmapped.pogo con rutas de acceso a la anotación GTF, secuencia de la proteína FASTA y péptido identificación archivo creado en el paso 3.2. También sustituir la MYSPECIES con la especie consistente con los datos (e.g., humanos).
- PoGo.exe - gtf \PATH\TO\GTF - fasta \PATH\TO\FASTA-en \PATH\TO\IN-formato de cama-especie MYSPECIES -mm 1
- Confirman ejecución de comando pulsando la tecla "Enter". Espere hasta que la ejecución haya terminado antes de progresar cualquier aún más.
Nota: Esto puede tardar unos minutos. El fichero resultante se almacenará en la misma carpeta que el archivo de entrada del péptido y se considerará como \PATH\TO\OUT.pogo_1MM.bed en el siguiente.
- Visualizar los péptidos asignados sin y con desajuste en el IGV como se describe en el paso 2.2.
4. mapeo usando múltiples archivos y la generación de ejes de pista para grandes conjuntos de datos
-
Mapeo de péptidos de varios archivos con PoGoGUI
- Desplácese a la carpeta de ejecutables e inicie el programa GUI mediante la ejecución de PoGoGUI-vX.X.X.jar.
- Seleccione el archivo ejecutable de PoGo para el sistema operativo en uso (aquí Linux), así como el archivo de referencia entrada proteína secuencias FASTA y el archivo GTF de anotación como se describe en los pasos de protocolo 2.1.2 - 2.1.4.
- Agregar los archivos de identificación de péptidos mediante el botón Agregar al lado de "Archivos de péptidos"; selección múltiple de archivos está habilitado, así como arrastrar y soltar en el campo en blanco debajo de "Archivos de péptidos".
- Desmarque las casillas de verificación junto a la cama de PTM, GTF y GCT en la sección de formatos de salida y sólo dejar cama comprobado.
- Seleccione la opción combinar archivos múltiples en una sola salida.
Nota: Esto resultará en un archivo de salida única combinando todos los péptidos de los archivos de entrada. Dejar esta opción sin seleccionar dará como resultado una ejecución secuencial del programa para cada archivo de entrada por separado. - Seleccionar las especies apropiadas para los datos de la lista desplegable selección consistente con los archivos FASTA y GTF.
- Inicio mapas haciendo clic en el botón Inicio . Si es necesario, el programa va a convertir los archivos de entrada formato de pogo. Esto podría tomar algún tiempo para ejecutar. Mientras tanto, descargar las herramientas necesarias y scripts para la generación de eje de pista.
-
Preparación para la generación de eje de pista
- Abra un navegador web, vaya a https://github.com/cschlaffner/TrackHubGenerator y descargar el archivo "TrackHubGenerator.pl". Guarde el archivo en la carpeta de ejecutables.
- En el explorador web, desplácese hasta www.hgdownload.soe.ucsc.edu/admin/exe/ y seleccione la carpeta para el sistema operativo en uso (Linux aquí). Descargue la herramienta bedToBigBed y el script fetchChromSizes en la carpeta de ejecutables27.
-
Generar un centro de la pista de péptidos asignadas
Nota: Después de que PoGoGUI haya terminado de asignar los péptidos, un centro de pista puede se generará automáticamente para todos los archivos resultantes en formato cama almacenada en la misma carpeta.- Abra una ventana de terminal y escriba el siguiente comando:
Perl TrackHubGenerator.pl PATH/a/nombre FBED UCSC correo electrónico- Sustituir nombre de ruta de a con una ruta de archivo y el nombre para el centro de la pista (por ejemplo, ~/PoGo/Data/Mytrackhub), conjunto con la Asamblea del genoma en que la anotación está basada (por ejemplo, hg38 para el ser humano), FBED con la ruta a la carpeta que contiene el Archivos de cama en que se basará el eje de pista (e.g., ~/PoGo/Data/), UCSC con la carpeta donde se almacenan las herramientas descargadas la UCSC (e.g., ~/PoGo/Executables/) y correo electrónico con una dirección de correo electrónico de la persona responsable de la pista concentrador.
- Confirmar la ejecución pulsando la tecla "Enter"; la ejecución sólo tendrá poco tiempo para terminar.
- Transferir el eje de pista generada (es decir, la carpeta creada ~/PoGo/Data/Mytrackhub/) con todo su contenido a un servidor FTP accesible desde la web.
Nota: Un servidor FTP con un servidor de web asociados que permite el acceso al centro de pista mediante los protocolos ftp y http se prefiere. Los repositorios github (github.com) y figshare (figshare.com) soporta este tipo de acceso y pueden ser utilizados en lugar de un servidor FTP.
- Abra una ventana de terminal y escriba el siguiente comando:
-
Visualizar un eje de pista en el browser del genoma UCSC
- En un explorador web, desplácese hasta https://genome.ucsc.edu/ y seleccione MyData | Seguimiento de centros. Haga clic en la ficha Mis ejes.
- Copiar la URL para el eje de pista en el campo de texto.
Nota: La URL consta de la dirección del servidor, la ubicación del eje de pista y nombre y el archivo de hub.txt (p. ej., http://ngs.sanger.ac.uk/production/proteogenomics/WTSI_proteomics_PandeyKusterCutler_tissues_hi/hub.txt). - Cargar el eje de la pista haciendo clic en Agregar Hub.
Nota: El cubo se cargan y aparece un mensaje corto, indicando detalles del centro de la pista como su nombre, la información de contacto de la persona responsable para el centro de la pista, y la Asamblea del genoma. La web volverá a la Página principal. - Seleccione GenomeBrowser para entrar en la vista de explorador.
Nota: El eje de la pista personalizada aparecerá en la parte superior de la lista. Si varios archivos de cama construcción la base para el centro de la pista, cada uno de los archivos se representará como una pista separada dentro del buje.
Resultados
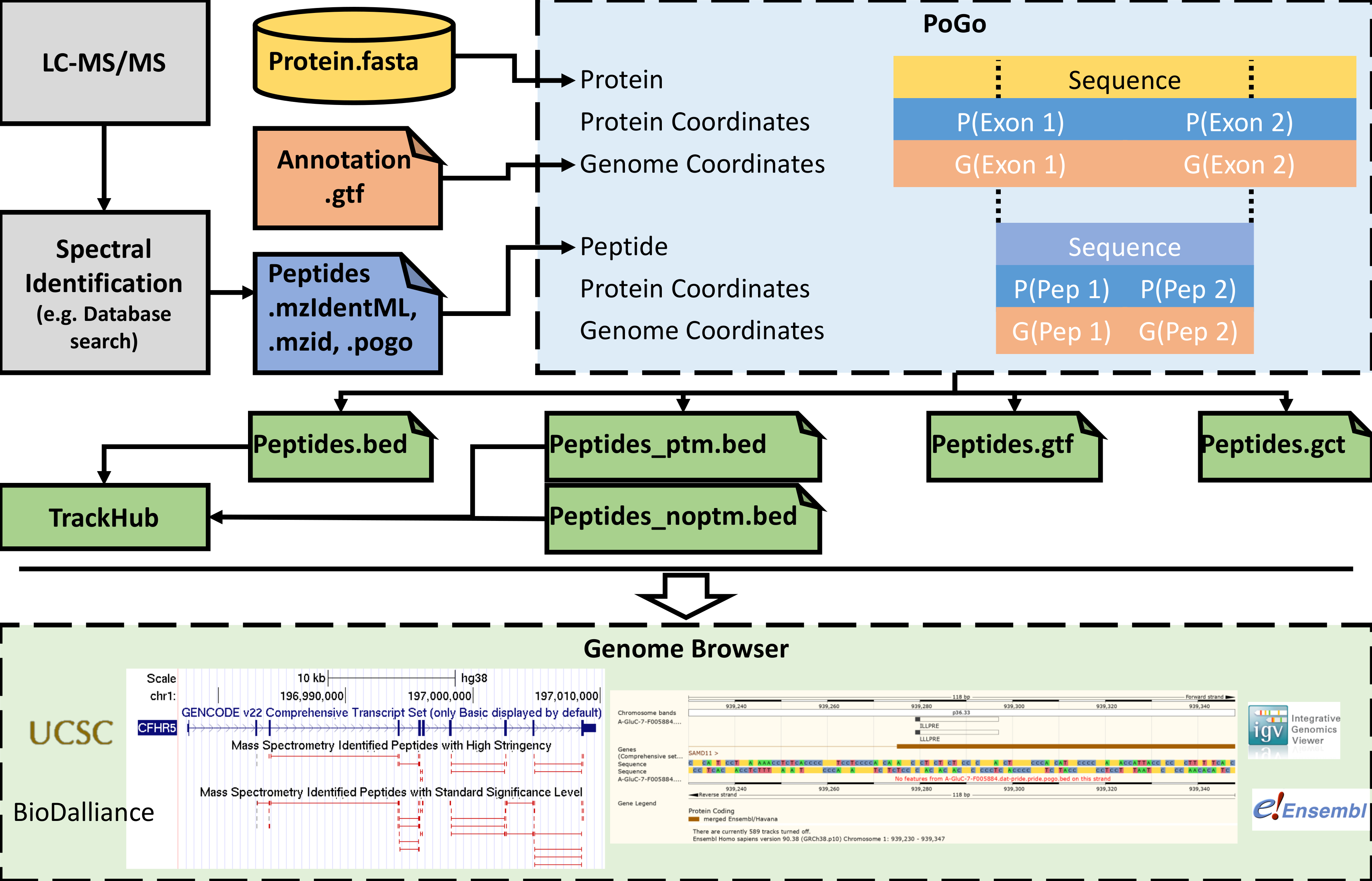
Una representación gráfica, resaltando en que etapa de un flujo de trabajo regular proteómicos PoGo18 se aplica, así como de aguas abajo opciones de visualización, se muestra en la figura 5. Proteomics de la escopeta (es decir, la digestión proteolítica de proteínas seguida de cromatografía de líquidos acompañada con espectrometría total en tándem) es un paso precursor de mapeo de proteogenomic. La espectrometría de masas tándem resultantes es comúnmente en comparación con espectros teóricos derivados de las bases de datos secuencia de proteína. Estudios de Proteogenomics introducen secuencias de traducción de novela transcripciones con la codificación de variantes de un solo nucleótido no sinónimo y potenciales (SNVs) en la base de datos, lo que hace difícil relacionarse fácilmente con estos detrás el genoma de referencia8. La interfaz gráfica de usuario de PoGo (PoGoGUI) soporta formatos de archivo de informes estandarizados de identificación de péptidos de los experimentos de espectrometría de masas y las convierte en el formato simplificado de 4 columna de pogo. PoGoGUI ajusta la herramienta de línea de comandos PoGo y así permite el mapeo de péptidos en coordenadas del genoma utilizando la anotación de referencia de codificación de la proteína los genes comúnmente proporcionado en el GTF y las secuencias de transcripción traducida en formato FASTA. Formatos de salida diferentes son generados por PoGo para permitir la visualización de los diferentes aspectos de los péptidos identificados a través de espectrometría de masas, incluyendo modificaciones post-traduccionales y cuantificación de niveles de péptido. Archivos de salida en la cama más pueden ser convertidos y combinados en directorios accesibles en línea llamados pista concentradores. Archivos de salida única, así como ejes de pista, entonces se pueden visualizar en navegadores como el Browser del genoma de UCSC25Ensembl genoma navegador20, IGV24y Biodalliance28 (ver figura 5 abajo).
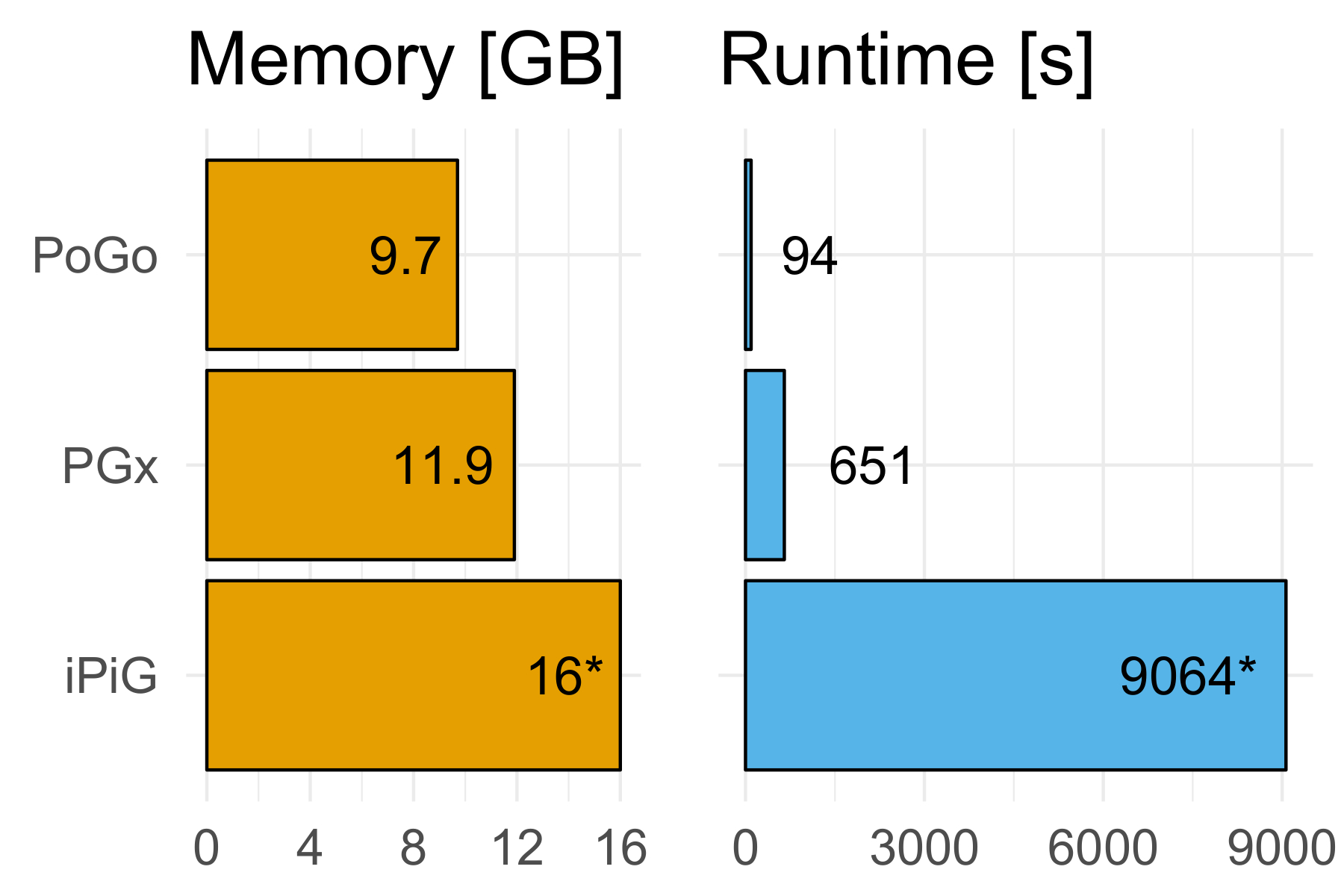
Aplicamos el PoGo para el reanálisis del proyecto proteoma humano mapas de filtrado en alta significación como se describe en Wright et al. 7 y respecto a dos otras herramientas para el mapeo de proteogenomic, a saber: iPiG14 y PGx10. El conjunto de datos compuesta por 233.055 péptidos únicos a través de 59 tejidos adultos y fetales, lo que resulta en un total de más 3 millones de secuencias. PoGo superó a estas herramientas en tiempo de ejecución (6,9 y 96.4 x más rápido, respectivamente) y uso de la memoria (20% y 60% menos de memoria, respectivamente) como se muestra en la figura 618. En la figura 7se muestra un ejemplo de un péptido con éxito asignado.
Mientras que PoGo superaron significativamente a las otras herramientas en velocidad y memoria, es también capaz de modificaciones poste-de translación de la cartografía y la información cuantitativa asociada a péptidos en el genoma. Figura 8A muestra esquemáticamente la visualización del formato de cama en un browser del genoma para péptidos traz a un exón y en empalme a ensambladuras. PoGo utiliza la opción de colorear para proporcionar fácil ayuda visual con respecto a la singularidad del mapeo de péptidos dentro del genoma. Asignaciones en rojo indican exclusividad a una sola transcripción, mientras que reflejos negro a un solo gen. Sin embargo, el péptido se comparte entre diferentes transcripciones. Asignaciones de gris muestran un péptido compartido entre múltiples genes. Estas son, por ejemplo, menos confiable para la cuantificación de un gen o llamar a la expresión de un gene. La opción de cama de PTM de PoGo redefine el código de colores para adaptarse a diferentes tipos de modificaciones post-traduccionales como se muestra en la figura 8B. Además, PTMs se indican por gruesos bloques (ver figura 8B). Un PTM solo de un tipo se destaca por un grueso bloque en la posición del residuo del aminoácido modificado, mientras que PTMs múltiples del mismo tipo están atravesados por un grueso bloque desde el primer aminoácido modificado a la última.
Aplicamos el PoGo y posteriormente TrackHubGenerator a un conjunto de datos de 50 líneas celulares de cáncer colorrectal como todo proteoma y phosphoproteome29. Mientras que el eje de pista cargado en el Browser del genoma de UCSC muestra los péptidos asignados al genoma y pone de relieve la singularidad de las asignaciones y los sitios de fosforilación (ver figura 9), datos adicionales se encuentran en la carpeta complementaria. Los archivos GCT luego activar la visualización de la cuantificación del péptido y fosfopéptidos en un contexto genómico. Sin embargo, los archivos GCT proporciona una visualización fácil de los péptidos que atraviesan a través de uniones de empalme (ver arriba figura 10 ). Los péptidos a través de uniones de empalme se dividen en sus partes respectivas a los exones. Si bien es posible identificar péptidos de empalme a través de los mismos valores cuantitativos de las asignaciones de exón, asignación de secuencia de carga archivos como cama o GTF que conectan los exones de un intrón delgado que soporte la interpretación (ver figura 10 parte inferior).
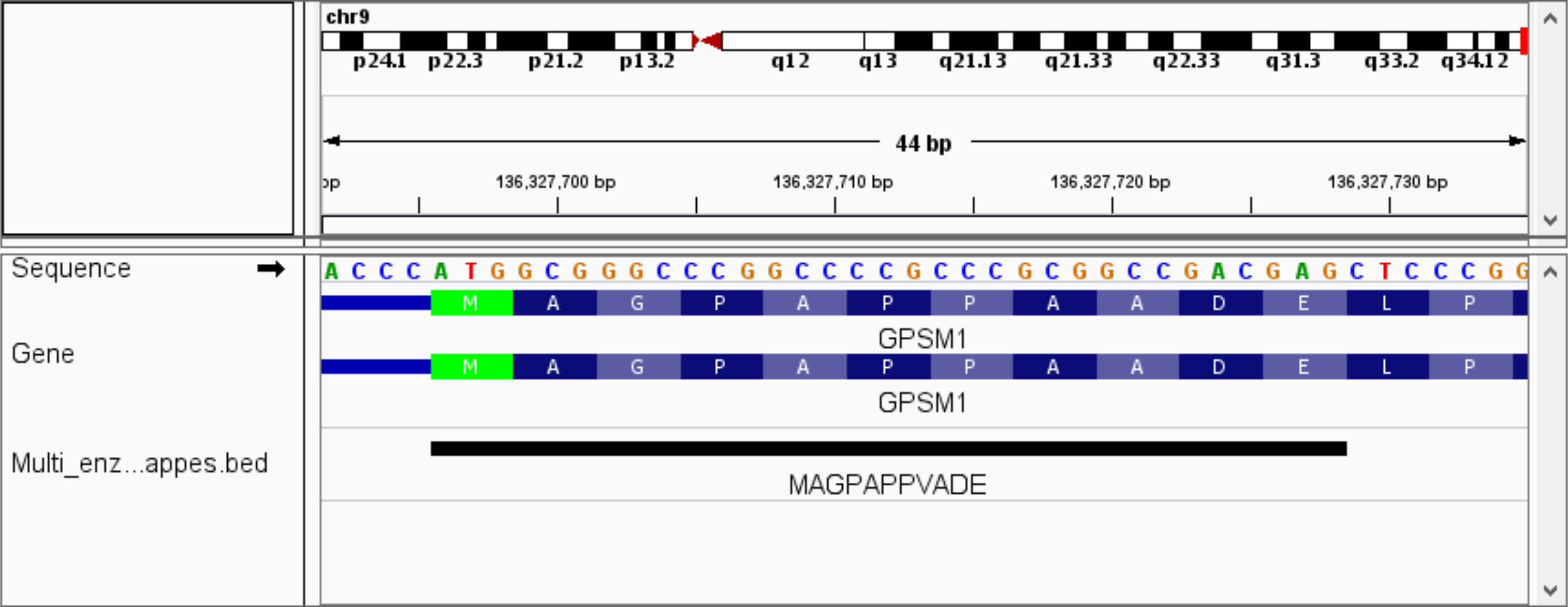
Para resaltar la utilidad de la variante con mapping, aplicamos PoGo en dos configuraciones a un conjunto de datos de proteoma humano testis búsquedas contra neXtProt a la caza de falta proteínas usando una estrategia de múltiples enzimas22. La neXtProt comprende además secuencias de la proteína de referencia sobre 5 millones de variantes solo aminoácido30. Mapeo de péptidos identificados con la variante de un solo aminoácido no es compatible con otras herramientas de mapeo. Se identificaron un total de 177.012 péptidos únicos. De estos péptidos 99.8% (176.694) primero fueron mapeadas con éxito sin permitir que las discrepancias. Quitar de la lista de péptido identificado dio lugar a péptidos de 0.2% (318) que posteriormente fueron asignadas permitiendo una substitución del aminoácido. Esto dio lugar a 3.446 asignaciones de 162 péptidos que no hubiera sido asignados al genoma de referencia con cualquier otra herramienta disponible. Mientras que el promedio de las asignaciones como una falta de coincidencia es alta, 62 péptidos fueron asignados a solamente un solo locus, indicando secuencias variante verdadera. Un ejemplo de un péptido con una sola substitución del aminoácido se destaca con su secuencia y la secuencia genomic traducida en la figura 11.

Figura 1. Comparación visual de herramientas de mapeo de péptidos a genoma diferentes. La comparación se muestra en varios aspectos. Estos aspectos incluyen una referencia de la cartografía, el grado de integración en los marcos y el apoyo de buscadores online y offline. Además, se destaca por separado aspectos novedosos de proteogenomics y su compatibilidad con la función. PoGo sólo carece de la capacidad para asignar directamente a una secuencia del genoma en comparación con otras herramientas. Sin embargo, es compatible con todas las características nuevas que no es compatibles con la mayoría de las otras herramientas. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2. Archivo de entrada de ejemplo de péptidos asignación. PoGo acepta datos de entrada en un formato separado por tabulador con 4 columnas. Encabezados de columna en la primera línea son 'Experimentar', 'Péptido', 'PSMs' y 'Quant', que indica en las siguientes líneas el experimento o identificador de la muestra, la secuencia del péptido, el número de péptido-espectro y un valor cuantitativo para el péptido, respectivamente. Extensiones de nombre de archivo compatibles son *.txt, *.tsv y *.pogo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3. Interfaz PoGoGUI con pasos resaltados para selecciones de archivo y opciones de parámetros. La figura muestra los pasos para seleccionar y cargar todos los archivos requeridos y la selección de opciones para péptidos de mapeo con modificaciones poste-de translación en el genoma humano de referencia. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4. Captura de pantalla de los datos del visor de genómica Integrativa (IGV) Añadir procedimiento. La figura destaca los pasos para subir archivos de salida de PoGo en el navegador IGV. Además, muestra la opción de ampliar la pista de péptidos asignadas para resaltar la asignación y la secuencia. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5. Simplificado de flujo de trabajo de medidas de LC-MS/MS para la visualización en navegadores de genoma. Asignación de PoGo sigue la identificación de péptidos de espectrometría de masas tándem. Para lograr el mapeo del genoma, PoGo utiliza la anotación de referencia como la anotación del genoma (GTF) y transcripción traducción secuencias (FASTA). Salida se generan formatos que se puede cargar por separado en los navegadores de genoma. Además, se pueden combinar archivos en formato cama en ejes de pista visualización de grandes conjuntos de datos de apoyo. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6. PoGo benchmarking contra PGx y iPiG. PoGo supera a las otras herramientas de evaluación comparativa. Mapeo de péptidos únicos 233.055 en 59 tejidos adultos y fetales, dando por resultado sobre 3 millones de secuencias, PoGo fue de 6,9 y 96.4 x más rápido que PGx y iPiG, respectivamente. Además, PoGo requerido 20% y 60% menos memoria en comparación con PGx y iPiG, respectivamente. Mientras que PoGo y PGx terminaron con éxito, iPiG dio lugar a un error de memoria de 16 GB. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7. Vista de ejemplo de explorador de genoma de UCSC de péptidos asignadas. La figura muestra péptidos asignados a la gen mTOR. Mientras que la pista combinada muestra los péptidos que atraviesan a través de uniones de empalme y asignación a un exón con las secuencias asociadas, las vías específicas de tejido sólo ponen de relieve la asignación en un formato condensado. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 8. Esquema de mapeo de visualización y codificación de color. (A) en el archivo de salida estándar de la cama, péptidos a un exón se muestran como bloques individuales (izquierdas), mientras que los péptidos mapeo a través de múltiples exones destaca el exón que abarca piezas como bloques (derecha). Intrones se muestran tan finos concatenación de líneas. PoGo color-codes la singularidad del trazado o péptidos y genes, transcritos usando un sistema de 3 niveles. (B) además de la estructura de bloque del formato de cama, cama de PTM salida destaca la posición de modificaciones post-traduccionales como bloques gruesos. La presencia de una PTM solo de un tipo destaca el residuo del aminoácido modificado con un espesor de bloque, mientras que varios sitios de la misma PTM se combinan en bloques de tiempo que abarca desde el primero hasta el último sitio de modificación. Asignaciones de péptidos se dividen por códec de tipo y color PTM, basado en la modificación. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 9. Seguimiento de centro vista en el navegador del genoma UCSC de cáncer colorrectal proteoma y phosphoproteome los datos. El centro de la pista compone de proteoma conjunto datos como phosphoproteome. Mientras que el color rojo en las pistas de proteoma y phosphoproteome indican la singularidad de la asignación a la sola transcripción de SFN, pistas en _ptm muestran los sitios de fosforilación en péptidos. Aquí, el color rojo indica el tipo de modificación como fosforilación. Sólo dos péptidos se han identificado con cada uno mostrando una única fosforilación (bloques de espesor). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 10. Vista de fosfotoproteida de cáncer colorrectal y cuantificación asociado en IGV. La figura muestra un subconjunto de las líneas celulares de 50 cáncer. Además muestra cuatro columnas de bloques en diferentes tonos de luz roja. El color indica la abundancia relativa de bajo (blanco) a alta (rojo). Mientras que las cuatro columnas inicialmente pueden llevar a creer que hay 4 péptidos, se hace evidente con el asociado basado en la secuencia GTF archivo de salida que de hecho son dos péptidos, que abarca a una ensambladura del empalme. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 11. Vista del péptido con variante de aminoácido en IGV. La figura muestra un péptido con una variante de aminoácido único asignada al genoma de referencia en el inicio de la traducción del gen GPSM1. La variante se encuentra en el residuo del aminoácido 8 y los resultados en la sustitución de alanina a valina (A→V). Las secuencias de la traducción de las transcripciones anotadas (azul) destacan la variante en comparación con la secuencia del péptido. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
Este protocolo describe cómo la herramienta de software PoGo y su interfaz gráfica de usuario PoGoGUI permiten un rápido mapeo de péptidos en coordenadas de genoma. La herramienta ofrece características únicas como modificación poste-de translación, cuantitativo y asignación basados en la variante de genomas mediante anotación de referencia. Este artículo muestra el método en un estudio a gran escala proteogenomic y destaca su eficiencia velocidad y memoria en comparación con otras herramientas disponibles18. En combinación con la herramienta TrackHubGenerator, que crea cubos en línea accesibles de genómica y genoma ligado de datos, PoGo, con una interfaz gráfica de usuario, estudios de proteogenomics a gran escala permite visualizar rápidamente sus datos en el contexto genómico. Además, se demuestran las características únicas del PoGo con conjuntos de datos de búsquedas en bases de datos variables y cuantitativa fosfoproteómico22,29.
Archivos individuales, como el archivo GCT, proporcionan visualización valiosa y relaciones entre características del péptido y loci genómicos. Sin embargo, es importante tener en cuenta que una interpretación basada en éstas solo puede ser difícil o engañoso debido a su limitación a solo aspectos de proteogenomics como singularidad, modificaciones post-traduccionales, valores cuantitativos. Por lo tanto, es importante elegir cuidadosamente que los archivos de salida, las opciones y combinaciones son apropiadas para la pregunta de proteogenomic en cuestión y modificar las combinaciones. Por ejemplo, información sobre la singularidad de la asignación a un locus genómico específico podría ser de gran valor para la anotación de una característica genómica7, mientras que la cuantificación en muestras diferentes puede ser más apropiada para estudios relacionados con el características genómicas a cambios en la abundancia de proteínas29. La salida debe ser generada por PoGo para cada ajuste. En caso de que no hay salida se genera, o archivos vacíos se muestran en la carpeta de salida, se recomienda que compruebe los archivos de entrada para el contenido deseado y el formato de archivo. En casos donde el contenido o formato de archivo no sigue las expectativas de PoGo (por ejemplo, el archivo FASTA que supuestamente contienen las secuencias de traducción transcripción contiene las secuencias de nucleótido de los transcritos), mensajes de error le preguntará al usuario Compruebe los archivos de entrada.
Las restricciones del protocolo y la herramienta en su mayoría se basan en la reutilización de los formatos de archivo utilizados en la genómica. Reasignación de formatos de archivo utilizados en la genómica para aplicaciones proteogenomic se acompaña de limitaciones específicas. Estas son debido a los diferentes conjuntos de requisitos para la visualización de genoma centrado de genómica y proteogenomic datos, como la necesidad de visualizar las modificaciones post-traduccionales de datos de proteómica. Esto se restringe en los formatos de archivo de genómica por el uso de una función. Han desarrollado muchos enfoques y herramientas de Proteómica con confianza localizar modificaciones post-traduccionales dentro de33,de péptido secuencias31,32,34. Sin embargo, la visualización de múltiples modificaciones de una manera única y discernible en el genoma es obstaculizada por la estructura de los formatos de archivo genómica. Por lo tanto, la visualización de bloque de PTMs múltiples del mismo tipo no constituye ninguna ambigüedad de los sitios de modificación pero es la consecuencia de la exigencia diferentes de la comunidad genómica sólo visualizar características individuales a la vez. Sin embargo, PoGo tiene la ventaja de modificaciones post-traduccionales de mapeo en coordenadas genómicas para permitir estudios centrados en el efecto de características genómicas como variantes de un solo nucleótido en modificaciones post-traduccionales. Con el PoGo, asignación variable aumenta el número de asignaciones total. Sin embargo, la codificación de color único de péptidos asignadas destaca asignaciones confiables de los poco fiables. El mapeo de péptidos variante identificada de variantes conocidas de un solo nucleótido puede acompañarse por visualizar los péptidos asignados junto a las variantes en formato VCF. De esta manera el código de color que indica una asignación poco fiable de un péptido variante es anulada por la presencia de la variante de nucleótidos conocido.
Un paso crítico para el uso de PoGo es el uso de los formatos y archivos correctos. El uso de secuencias de transcripción traducida como secuencias de la proteína para acompañar a la anotación en formato GMT es el criterio principal. Otro elemento crítico cuando se considera usar PoGo a péptidos con aminoácidos desajustes es memoria. Mientras que memoria eficientes para una aplicación estándar, significativamente y exponencialmente creciente número de posibles asignaciones con uno o dos desajustes conduce a un aumento igualmente exponencial en el uso de memoria18. Se propone una asignación de etapas como se describe en este protocolo primero los péptidos sin desajustes y quitar del conjunto. Los péptidos previamente asignados posterior entonces pueden asignarse mediante un desajuste y el procedimiento puede repetirse con dos desajustes para los péptidos restante sin asignar.
Puesto que el rendimiento de la espectrometría de masas ha aumentado significativamente y estudios interconexión genómicos y proteómicos son cada vez más frecuentes en los últimos años, son herramientas que permiten fácilmente interfaces estos tipos de datos en el mismo sistema de coordenadas cada vez más indispensable. La herramienta presentada aquí le ayudará a la necesidad de combinar genómica y los datos proteómicos para potenciar un mejor entendimiento de estudios integrados a través de pequeños y grandes conjuntos de datos mediante la asignación de péptidos en una anotación de referencia. Es alentador, PoGo se ha aplicado para asignar péptidos a los candidatos del gen en el mismo formato que la anotación de referencia para apoyar los esfuerzos de la anotación de genes nuevos en testículo humano35. El enfoque presentado aquí es independiente de bases de datos utilizadas para la identificación de péptidos. El protocolo podría ayudar en la identificación y visualización de los productos de la traducción de novela mediante el uso de había adaptado entrados archivos de secuencias de traducción y asociados archivos GTF de RNA-seq experimentos.
Varios enfoques y herramientas con una amplia gama de escenarios de aplicación especial para asignar coordenadas genómicas, desde el mapeo de péptidos directamente a la secuencia del genoma a los mapas de secuencia de RNA guiada, péptidos han sido introducidas10, 11 , 12 , 13 , 14 , 15 , 16 , 17. sin embargo, estos pueden resultar en un fracaso para asignar correctamente péptidos cuando existen modificaciones post-traduccionales y errores en el mapa subyacente de Lee de la secuencia de RNA pueden ser propagados hasta el nivel de péptido. PoGo se ha desarrollado específicamente superar esos obstáculos y hacer frente con el rápido aumento de conjuntos de datos de proteómica cuantitativa de alta resolución para integrar con plataformas de genómica ortogonal. La herramienta descrita aquí puede integrarse en flujos de trabajo de alto rendimiento. A través de la interfaz gráfica de PoGoGUI, la herramienta es fácil de usar y no requiere especialista en Bioinformática formación.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Este trabajo fue financiado por el Wellcome Trust (WT098051) y la subvención del NIH (U41HG007234) para el proyecto GENCODE.
Materiales
| Name | Company | Catalog Number | Comments |
| PoGo (software) | NA | NA | https://github.com/cschlaffner/PoGo |
| PoGoGUI (software) | NA | NA | https://github.com/cschlaffner/PoGoGUI |
| TrackHubGenerator (software) | NA | NA | https://github.com/cschlaffner/TrackHubGenerator |
| Integrative Genomics Viewer (software) | NA | NA | http://software.broadinstitute.org/software/igv/ |
| UCSC genome browser (website) | NA | NA | https://genome.ucsc.edu/ |
| GENCODE (website) | NA | NA | http://gencodegenes.org |
| Ensembl (website) | NA | NA | http://ensembl.org |
| bedToBigBed (software) | NA | NA | http://hgdownload.soe.ucsc.edu/admin/exe/ |
| fetchChromSizes.sh (software) | NA | NA | http://hgdownload.soe.ucsc.edu/admin/exe/ |
Referencias
- Aebersold, R., Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature. 537 (7620), 347-355 (2016).
- Mertins, P., et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 534 (7605), 55-62 (2016).
- Zhang, H., et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 166 (3), 755-765 (2016).
- Jaffe, J. D., Berg, H. C., Church, G. M. Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics. 4 (1), 59-77 (2004).
- Wilhelm, M., et al. Mass-spectrometry-based draft of the human proteome. Nature. 509 (7502), 582-587 (2014).
- Kim, M. S., et al. A draft map of the human proteome. Nature. 509 (7502), 575-581 (2014).
- Wright, J. C., et al. Improving GENCODE reference gene annotation using a high-stringency proteogenomics workflow. Nature Communications. 7, 11778 (2016).
- Nesvizhskii, A. I. Proteogenomics: concepts, applications and computational strategies. Nature Methods. 11 (11), 1114-1125 (2014).
- Armengaud, J., et al. Non-model organisms, a species endangered by proteogenomics. Journal of Proteomics. 105, 5-18 (2014).
- Askenazi, M., Ruggles, K. V., Fenyo, D. PGx: putting peptides to BED. Journal of Proteome Research. 15 (3), 795-799 (2016).
- Choi, S., Kim, H., Paek, E. ACTG: novel peptide mapping onto gene models. Bioinformatics. 33 (8), 1218-1220 (2017).
- Ghali, F., et al. ProteoAnnotator-open source proteogenomics annotation software supporting PSI standards. Proteomics. 14 (23-24), 2731-2741 (2014).
- Has, C., Lashin, S. A., Kochetov, A. V., Allmer, J. PGMiner reloaded, fully automated proteogenomic annotation tool linking genomes to proteomes. Journal of Integrative Bioinformatics. 13 (4), 293 (2016).
- Kuhring, M., Renard, B. Y. iPiG: integrating peptide spectrum matches into genome browser visualizations. PLoS One. 7 (12), e50246 (2012).
- Pang, C. N., et al. Tools to covisualize and coanalyze proteomic data with genomes and transcriptomes: validation of genes and alternative mRNA splicing. Journal of Proteome Research. 13 (1), 84-98 (2014).
- Sanders, W. S., et al. The proteogenomic mapping tool. BMC Bioinformatics. 12 (115), (2011).
- Wang, X., et al. ProBAMsuite, a bioinformatics framework for genome-based representation and analysis of proteomics data. Molecular & Cellular Proteomics. 15 (3), 1164-1175 (2016).
- Schlaffner, C. N., Pirklbauer, G. J., Bender, A., Choudhary, J. S. Fast, quantitative and variant enabled mapping of peptides to genomes. Cell Systems. 5 (2), 152-156 (2017).
- Vizcaino, J. A., et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Research. 41, D1063-D1069 (2013).
- Aken, B. L., et al. Ensembl 2017. Nucleic Acids Research. 45 (D1), D635-D642 (2017).
- Perez-Riverol, Y., et al. Ms-data-core-api: an open-source, metadata-oriented library for computational proteomics. Bioinformatics. 31 (17), 2903-2905 (2015).
- Wang, Y., et al. Multi-protease strategy identifies three PE2 missing proteins in human testis tissue. Journal of Proteome Research. , (2017).
- Greseth, M. D., Carter, D. C., Terhune, S. S., Traktman, P. Proteomic screen for cellular targets of the vaccinia virus F10 protein kinase reveals that phosphorylation of mDia regulates stress fiber formation. Molecular & Cellular Proteomics. 16 (4 Suppl 1), S124-S143 (2017).
- Thorvaldsdottir, H., Robinson, J. T., Mesirov, J. P. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 14 (2), 178-192 (2013).
- Kent, W. J., et al. The human genome browser at UCSC. Genome Research. 12 (6), 996-1006 (2002).
- The R Development Core Team. . R: A Language and Environment for Statistical Computing. , (2008).
- Kent, W. J., Zweig, A. S., Barber, G., Hinrichs, A. S., Karolchik, D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 26 (17), 2204-2207 (2010).
- Down, T. A., Piipari, M., Hubbard, T. J. Dalliance: interactive genome viewing on the web. Bioinformatics. 27 (6), 889-890 (2011).
- Roumeliotis, T. I., et al. Genomic determinants of protein abundance variation in colorectal cancer cells. Cell Reports. 20 (9), 2201-2214 (2017).
- Gaudet, P., et al. The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Research. 45, D177-D182 (2017).
- Fermin, D., Walmsley, S. J., Gingras, A. C., Choi, H., Nesvizhskii, A. I. LuciPHOr: algorithm for phosphorylation site localization with false localization rate estimation using modified target-decoy approach. Molecular & Cellular Proteomics. 12 (11), 3409-3419 (2013).
- Fermin, D., Avtonomov, D., Choi, H., Nesvizhskii, A. I. LuciPHOr2: site localization of generic post-translational modifications from tandem mass spectrometry data. Bioinformatics. 31 (7), 1141-1143 (2015).
- Hansen, T. A., Sylvester, M., Jensen, O. N., Kjeldsen, F. Automated and high confidence protein phosphorylation site localization using complementary collision-activated dissociation and electron transfer dissociation tandem mass spectrometry. Analytical Chemistry. 84 (22), 9694-9699 (2012).
- Taus, T., et al. Universal and confident phosphorylation site localization using phosphoRS. Journal of Proteome Research. 10 (12), 5354-5362 (2011).
- Weisser, H., Wright, J. C., Mudge, J. M., Gutenbrunner, P., Choudhary, J. S. Flexible data analysis pipeline for high-confidence proteogenomics. Journal of Proteome Research. 15 (12), 4686-4695 (2016).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados